Clear Sky Science · nl
Krachtvrije moleculaire dynamica via autoregressieve equivariantie-netwerken
Waarom het versnellen van atomaire films ertoe doet
Veel van de meest intrigerende vragen in de scheikunde, natuurkunde en materialenwetenschap draaien om hoe atomen bewegen: hoe een pijnstiller-molecuul wiebelt, hoe kwarts trilt, hoe water bevriest tot vreemde glasachtige vormen. Computerexperimenten, moleculaire dynamica genoemd, simuleren deze atomaire films, maar ze zijn zo rekenintensief dat onderzoekers vaak slechts een klein deel van de actie zien. Dit artikel presenteert TrajCast, een nieuw kunstmatig-intelligentie-kader dat leert deze atomaire films vooruit te spoelen zonder de essentiële fysica op te offeren.
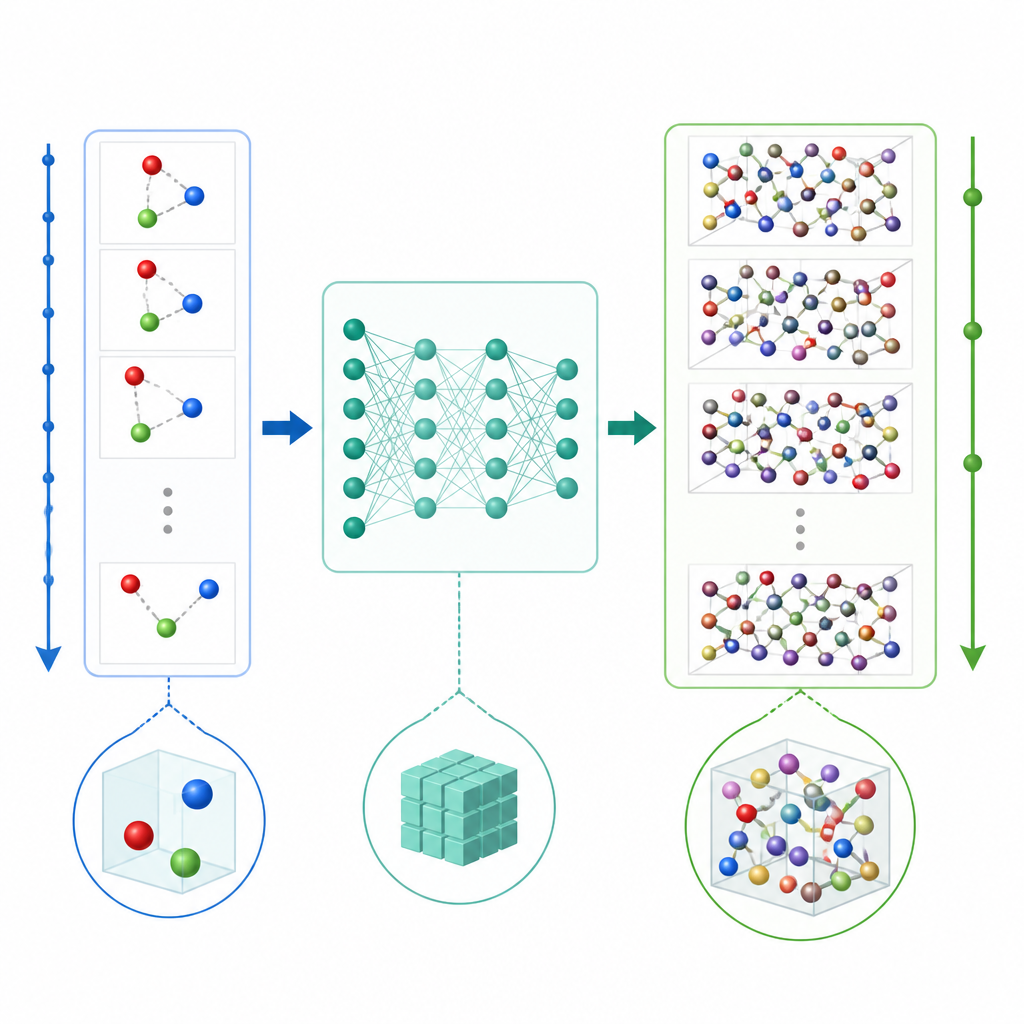
Een nieuwe snelkoppeling voor het simuleren van atomen
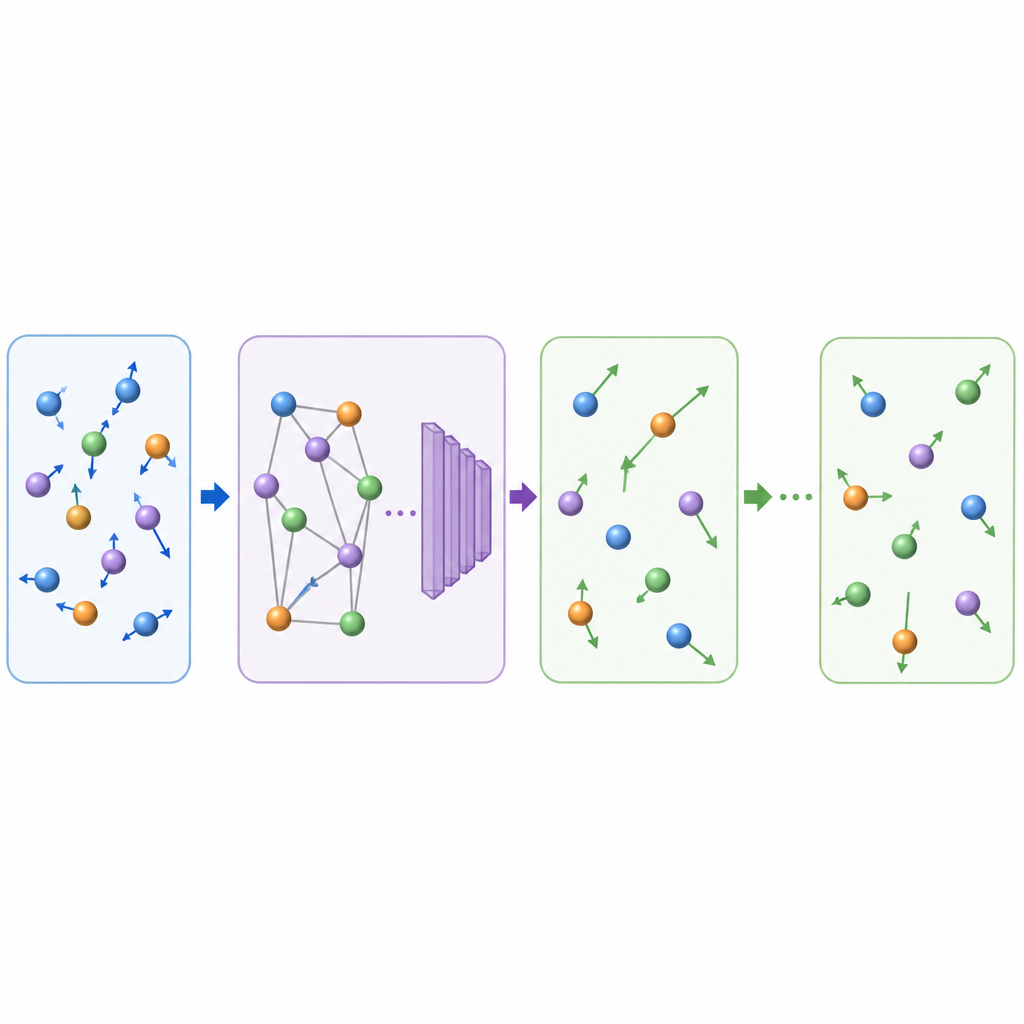
Traditionele moleculaire dynamica werkt atomaire bewegingen bij door herhaaldelijk de krachten tussen atomen te berekenen en vervolgens de posities in zeer kleine tijdstappen vooruit te schuiven, vaak een fractie van een biljoenste van een seconde. Zelfs wanneer machine learning wordt gebruikt om de krachtberekeningen te versnellen, maakt de minuscule tijdstap lange films nog steeds duur. TrajCast kiest een andere route. In plaats van krachten te voorspellen, leert het direct van de ene volledige snapshot van het systeem naar de volgende te springen, waarbij het zowel voorspelt waar elk atoom zal zijn als hoe snel het zal bewegen na een veel groter tijdsinterval. Onder de motorkap gebruikt TrajCast een klasse neurale netwerken die de symmetrieën van driedimensionale ruimte respecteren, zodat het roteren of transleren van het systeem het model niet in de war brengt.
De AI leren fysieke regels te respecteren
Om als een betrouwbare plaatsvervanger voor de fysica te fungeren, moet TrajCast meer doen dan plausibele plaatjes tekenen; het moet de statistieken van echte atomaire beweging reproduceren. De auteurs trainen het model op korte, nauwkeurige simulaties die energie en impuls behouden, en ze bouwen deze behoudswetten direct in het netwerk zodat de voorspelde bewegingen fysisch consistent blijven. Tijdens lange "roll-outs" neemt TrajCast herhaaldelijk zijn eigen output als volgende input, terwijl een thermostaatmodule de atomaire snelheden zachtjes bijstuurt om de gesimuleerde temperatuur onder controle te houden, net zoals in standaard simulaties bij constante temperatuur. Dit ontwerp maakt het mogelijk volledige trajecten te genereren waaruit onderzoekers dynamische, structurele en energetische eigenschappen kunnen halen.
TrajCast op de proef stellen
Het team evalueert TrajCast op drie zeer verschillende systemen: een enkel paracetamol-molecuul in de gasfase, kristallijne kwarts en vloeibaar water bij kamertemperatuur. Voor elk geval vergelijken ze de door AI gegenereerde trajecten met conventionele simulaties, met aandacht voor grootheden zoals hoe atomen trillen, hoe energie verdeeld is en hoe moleculen bewegen of diffusie vertonen. Voor paracetamol gebruikt TrajCast tijdstappen veertien keer groter dan in de referentiesimulatie, en toch reproduceert het nauwkeurig het spectrum van vibrationale bewegingen, de spreiding van potentiële energieën en het energielandschap dat bepaalt hoe het molecuul tussen vormen omslaat. In kwarts kan het veilig sprongen maken die dertig keer groter zijn dan gebruikelijk, terwijl het toch de trillingskenmerken en energiefluitingen van de vaste stof over nanoseconden evenaart.
Van vloeibaar water naar glasachtige toestanden
Vloeibaar water vormt een zwaardere uitdaging omdat moleculen voortdurend waterstofbruggen vormen en verbreken. Zelfs hier, met een tienmaal grotere tijdstap, weerspiegelt TrajCast de referentiesimulaties: het vangt vibratiemodi over frequenties, reproduceert hoe ver watermoleculen zich over tijd verplaatsen en levert radiale verdelingsfuncties die beschrijven hoe moleculen rondom elkaar zijn gerangschikt. De auteurs duwen het model vervolgens ver buiten zijn trainingscomfortzone. Met een versie die alleen op vloeibaar water bij kamertemperatuur is getraind, koelen ze geleidelijk een groter monster af om hypergequenched glasachtig water te vormen, een ongeordende vaste stof met subtiele structurele kenmerken. Opmerkelijk genoeg volgt de AI hoe diffusie vertraagt, hoe structurele pieken verscherpen en hoe maten van lange- en kortafstandordening evolueren, in nauwe overeenstemming met toegewijde simulaties die expliciet afkoeling omvatten.
Wat dit betekent voor toekomstige materiaalontdekking
Door te leren atomaire films in grotere, fysisch getrouwe sprongen vooruit te brengen, kan TrajCast nanoseconden aan trajectdata genereren voor systemen met duizenden atomen in de tijd die normaal nodig is om veel minder te simuleren. De methode behoudt voldoende detail om sleutelobservabelen terug te winnen, verwerkt zowel eenvoudige moleculen als complexe gecondenseerde fasen, en kan zelfs extrapoleren naar nieuwe regimes zoals glasvorming zonder extra training. Hoewel de huidige versie nog geen drukafhankelijke eigenschappen kan voorspellen en een vaste voorspellingsstap gebruikt, wijst het al op een toekomst waarin AI-ondersteunde simulaties de kloof overbruggen tussen snelle microscopische bewegingen en de langzame processen die materialen en vloeistoffen in de echte wereld vormen.
Bronvermelding: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Trefwoorden: moleculaire dynamica, machine learning, atomistische simulatie, materialenwetenschap, glasachtig water