Clear Sky Science · es
Dinámica molecular sin fuerzas mediante redes autorregresivas equivariantes
Por qué importa acelerar las películas atómicas
Muchas de las preguntas más intrigantes en química, física y ciencia de materiales se reducen a cómo se mueven los átomos: cómo se retuerce una molécula analgésica, cómo vibra el cuarzo, cómo el agua se congela en formas vítreas extrañas. Experimentos computacionales llamados dinámica molecular simulan estas películas atómicas, pero son tan exigentes computacionalmente que los científicos a menudo solo ven una fracción mínima de la acción. Este artículo presenta TrajCast, un nuevo marco de inteligencia artificial que aprende a avanzar rápidamente estas películas atómicas sin sacrificar la física esencial.
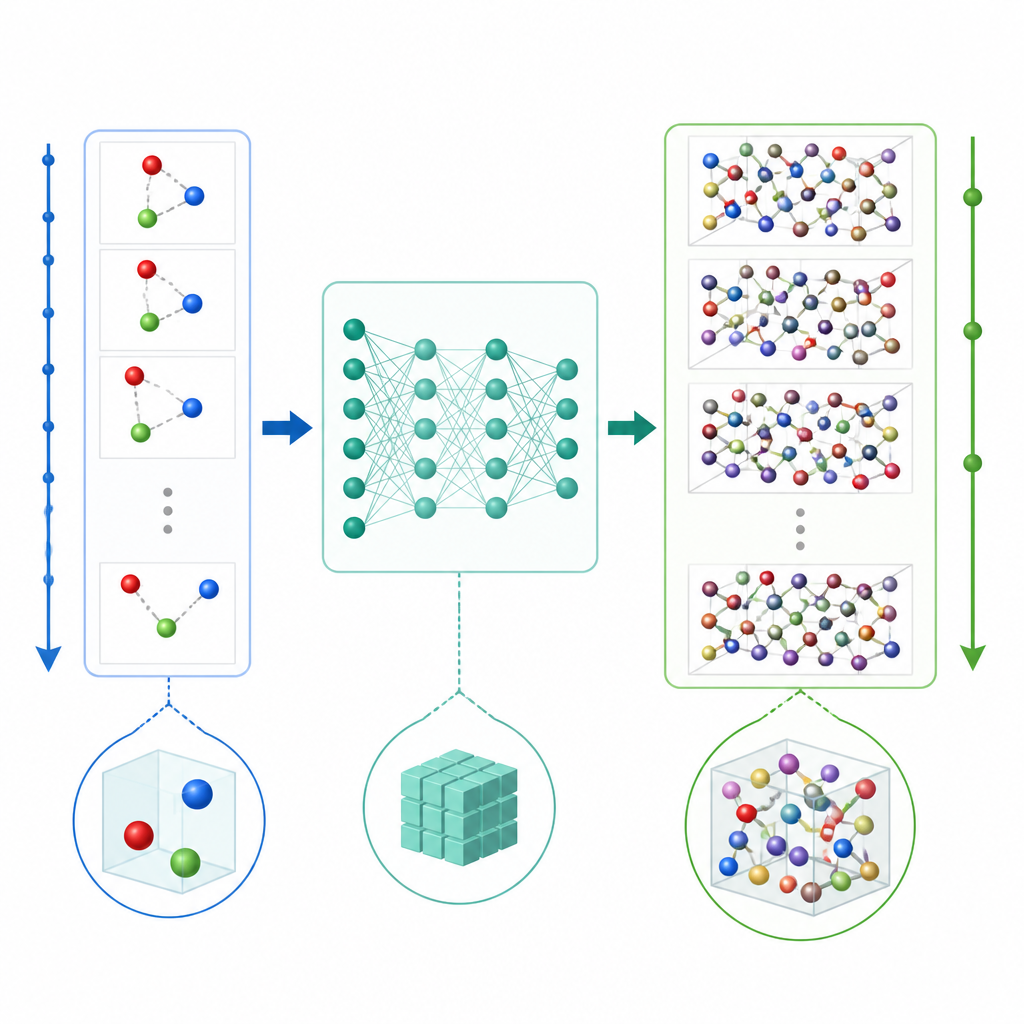
Un nuevo atajo para simular átomos
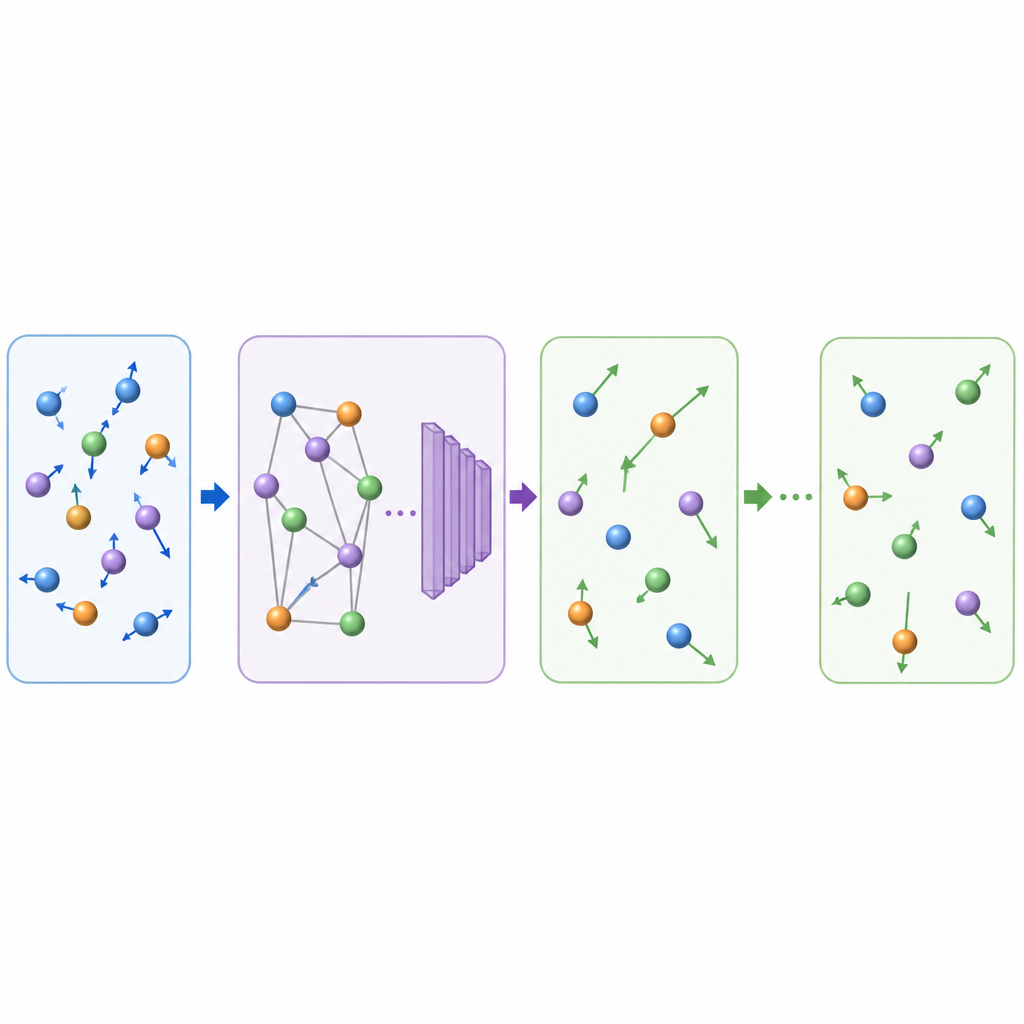
La dinámica molecular tradicional actualiza el movimiento atómico calculando repetidamente las fuerzas entre átomos y luego desplazando las posiciones hacia adelante en pasos de tiempo muy pequeños, a menudo una fracción de un billonésimo de segundo. Incluso cuando se usa aprendizaje automático para acelerar el cálculo de fuerzas, el diminuto paso temporal sigue haciendo que las películas largas sean costosas. TrajCast toma una ruta diferente. En lugar de predecir fuerzas, aprende a saltar directamente de una instantánea completa del sistema a la siguiente, pronosticando tanto dónde estará cada átomo como qué rapidez tendrá después de un intervalo de tiempo mucho mayor. Bajo el capó, TrajCast utiliza una clase de redes neuronales que respetan las simetrías del espacio tridimensional, de modo que rotar o trasladar el sistema no confunde al modelo.
Enseñar a la IA a respetar las leyes físicas
Para actuar como un sustituto fiable de la física, TrajCast debe hacer más que dibujar imágenes plausibles; debe reproducir las estadísticas del movimiento atómico real. Los autores entrenan el modelo con simulaciones cortas y precisas que conservan la energía y el momento, e incorporan estas leyes de conservación directamente en la red para que los movimientos predichos permanezcan consistentes físicamente. Durante los largos «roll‑outs», TrajCast toma repetidamente su propia salida como la siguiente entrada, mientras un módulo termostato ajusta suavemente las velocidades atómicas para mantener la temperatura simulada bajo control, tal como en las simulaciones estándar a temperatura constante. Este diseño permite que el método genere trayectorias completas de las que los científicos pueden extraer propiedades dinámicas, estructurales y energéticas.
Poniendo a prueba a TrajCast
El equipo evalúa TrajCast en tres sistemas muy diferentes: una sola molécula de paracetamol en fase gaseosa, cuarzo cristalino y agua líquida en condiciones ambientales. Para cada caso, comparan las trayectorias generadas por la IA con simulaciones convencionales, enfocándose en medidas como cómo vibran los átomos, cómo se distribuye la energía y cómo se mueven o difunden las moléculas. Para el paracetamol, TrajCast utiliza pasos temporales catorce veces mayores que la simulación de referencia, y aun así reproduce con fidelidad el espectro de movimientos vibracionales, la distribución de energías potenciales y el paisaje de barreras energéticas que gobiernan cómo la molécula cambia entre conformaciones. En el cuarzo, puede dar con seguridad saltos treinta veces mayores de lo habitual, manteniendo las firmas vibratorias y las fluctuaciones energéticas del sólido a lo largo de nanosegundos.
Del agua líquida a los estados vítreos
El agua líquida plantea un desafío más duro porque las moléculas forman y rompen constantemente enlaces de hidrógeno. Incluso aquí, con un paso temporal diez veces mayor, TrajCast refleja las simulaciones de referencia: captura modos vibracionales a lo largo de las frecuencias, reproduce cuánto viajan las moléculas de agua con el tiempo y ofrece funciones de distribución radial que describen cómo se disponen las moléculas unas respecto a otras. Los autores luego empujan el modelo mucho más allá de su zona de confort de entrenamiento. Usando una versión entrenada solo con agua líquida a temperatura ambiente, enfrían gradualmente una muestra mayor para formar agua vítrea hiperquenada, un sólido desordenado con firmas estructurales sutiles. De forma notable, la IA sigue cómo se ralentiza la difusión, cómo se acentúan picos estructurales y cómo evolucionan medidas de orden de largo y corto alcance, en estrecha concordancia con simulaciones dedicadas que incluyen explícitamente el enfriamiento.
Qué significa esto para el descubrimiento futuro de materiales
Al aprender a avanzar las películas atómicas en saltos mayores y físicamente fieles, TrajCast puede generar nanosegundos de datos de trayectoria para sistemas que contienen miles de átomos en el tiempo que normalmente tomaría simular mucho menos. El método conserva suficiente detalle para recuperar observables clave, maneja tanto moléculas simples como fases condensadas complejas, e incluso puede extrapolar a nuevos regímenes como la formación de vidrio sin entrenamiento adicional. Aunque la versión actual aún no puede predecir propiedades dependientes de la presión y usa un paso de predicción fijo, ya apunta a un futuro en el que las simulaciones asistidas por IA salten la brecha entre los rápidos movimientos microscópicos y los procesos lentos que moldean materiales y líquidos en el mundo real.
Cita: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Palabras clave: dinámica molecular, aprendizaje automático, simulación atomística, ciencia de materiales, agua vítrea