Clear Sky Science · fr
Dynamiques moléculaires sans forces grâce à des réseaux équivariants autorégressifs
Pourquoi accélérer les « films » atomiques est crucial
Beaucoup des questions les plus captivantes en chimie, physique et science des matériaux se ramènent au mouvement des atomes : comment une molécule d’antidouleur se tortille, comment le quartz vibre, comment l’eau gèle en formes vitreuses étranges. Les expériences numériques appelées dynamiques moléculaires simulent ces films atomiques, mais elles sont si coûteuses en calcul que les scientifiques n’en observent souvent qu’une infime partie. Cet article présente TrajCast, un nouveau cadre d’intelligence artificielle qui apprend à accélérer ces films atomiques sans sacrifier la physique essentielle.
Un nouveau raccourci pour simuler les atomes
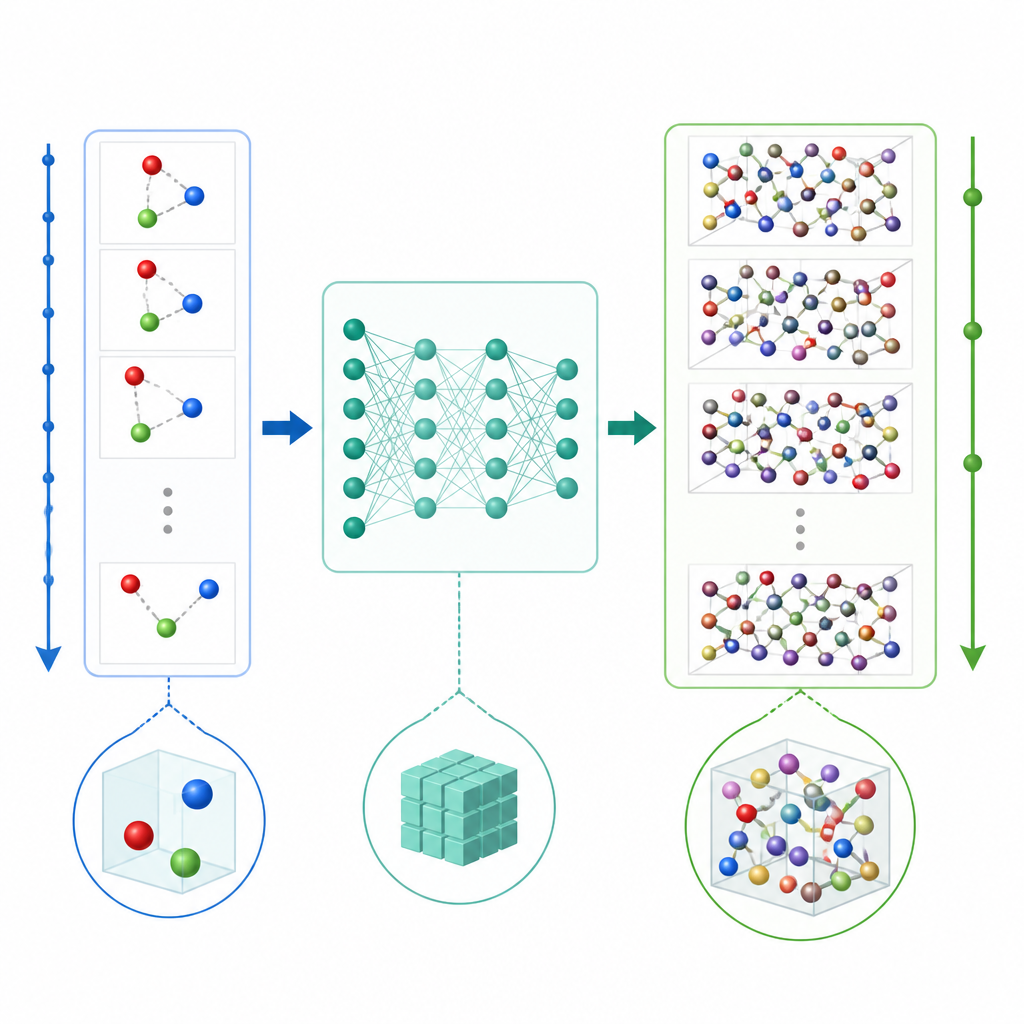
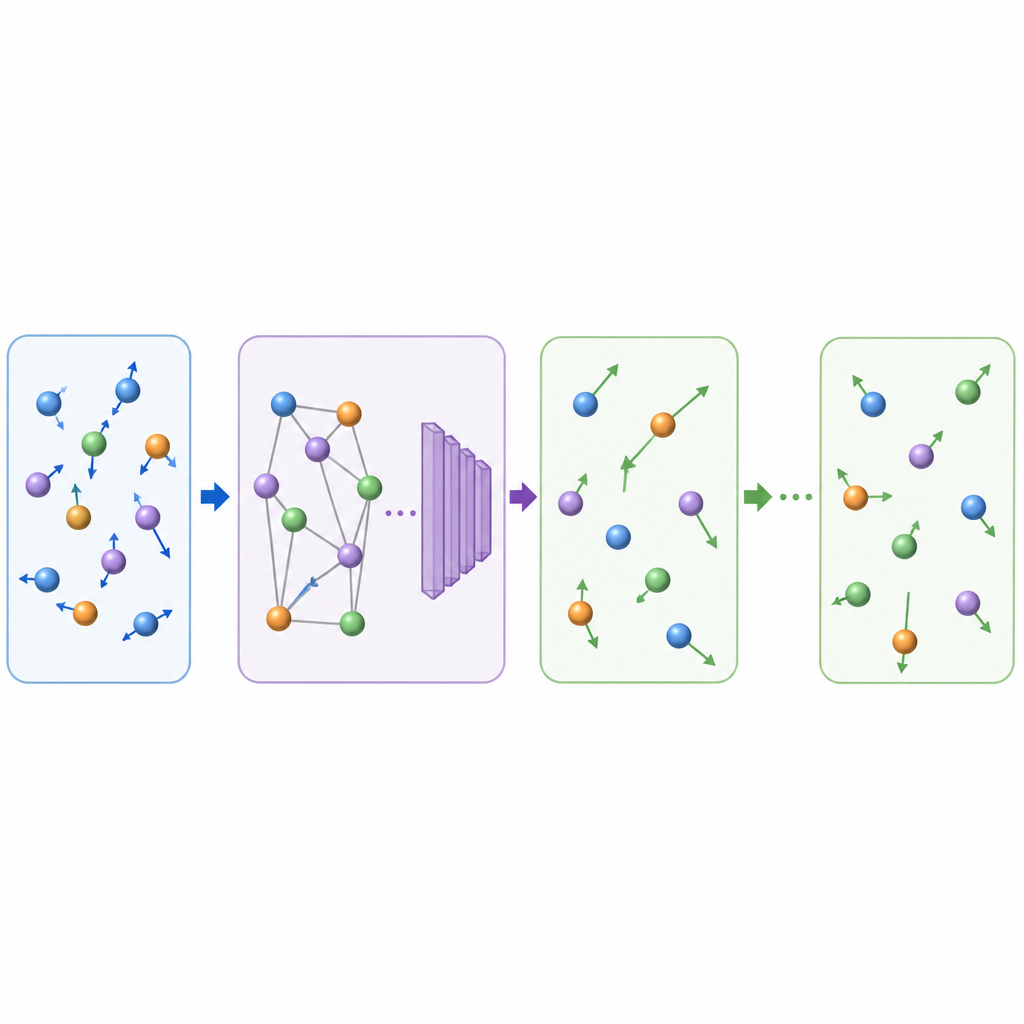
Les dynamiques moléculaires traditionnelles mettent à jour le mouvement atomique en calculant à répétition les forces entre atomes puis en avançant les positions par de très petits pas de temps, souvent une fraction de trillionième de seconde. Même lorsque l’apprentissage automatique accélère le calcul des forces, le petit pas de temps rend les longs films coûteux. TrajCast emprunte une voie différente. Plutôt que de prédire les forces, il apprend à passer directement d’un instantané complet du système au suivant, en prévoyant à la fois où chaque atome se trouvera et à quelle vitesse il se déplacera après un intervalle de temps beaucoup plus grand. Sous le capot, TrajCast utilise une classe de réseaux neuronaux qui respecte les symétries de l’espace tridimensionnel, de sorte que la rotation ou la translation du système ne perturbe pas le modèle.
Apprendre à l’IA à respecter les lois physiques
Pour jouer un rôle de substitut fiable à la physique, TrajCast doit faire plus que dessiner des images plausibles ; il doit reproduire les statistiques du mouvement atomique réel. Les auteurs entraînent le modèle sur des simulations courtes et précises qui conservent l’énergie et la quantité de mouvement, et ils incorporent ces lois de conservation directement dans le réseau afin que les mouvements prédits restent cohérents physiquement. Lors de longs « déroulés » (roll‑outs), TrajCast reprend de manière itérative sa propre sortie comme entrée suivante, tandis qu’un module thermostat ajuste en douceur les vitesses atomiques pour maintenir la température simulée sous contrôle, comme dans les simulations standard à température constante. Cette conception permet à la méthode de générer des trajectoires complètes à partir desquelles les scientifiques peuvent extraire des propriétés dynamiques, structurelles et énergétiques.
Mettre TrajCast à l’épreuve
L’équipe évalue TrajCast sur trois systèmes très différents : une seule molécule de paracétamol en phase gazeuse, du quartz cristallin et de l’eau liquide aux conditions ambiantes. Pour chaque cas, ils comparent les trajectoires générées par l’IA avec des simulations conventionnelles, en se concentrant sur des mesures telles que la façon dont les atomes vibrent, la répartition de l’énergie et le mouvement ou la diffusion des molécules. Pour le paracétamol, TrajCast utilise des pas de temps quatorze fois plus grands que la simulation de référence, et reproduit néanmoins fidèlement le spectre des mouvements vibratoires, la distribution des énergies potentielles et le paysage des barrières énergétiques qui gouvernent la bascule entre conformations. Dans le quartz, il peut prendre en toute sécurité des sauts trente fois plus grands que d’habitude, tout en reproduisant les signatures vibratoires et les fluctuations d’énergie du solide sur des nanosecondes.
De l’eau liquide aux états vitreux
L’eau liquide présente un défi plus difficile parce que les molécules forment et rompent continuellement des liaisons hydrogène. Même ici, avec un pas de temps dix fois plus grand, TrajCast reflète les simulations de référence : il capture des modes vibratoires sur une large gamme de fréquences, reproduit la distance parcourue par les molécules d’eau au fil du temps et fournit des fonctions de distribution radiale décrivant l’organisation des molécules les unes par rapport aux autres. Les auteurs poussent ensuite le modèle bien au‑delà de sa zone de confort d’entraînement. En utilisant une version entraînée uniquement sur l’eau liquide à température ambiante, ils refroidissent progressivement un échantillon plus grand pour former de l’eau vitreuse hyper‑refroidie, un solide désordonné aux signatures structurelles subtiles. De façon remarquable, l’IA suit le ralentissement de la diffusion, l’accentuation des pics structurels et l’évolution des mesures d’ordre à courte et longue portée, en bon accord avec des simulations dédiées qui incluent explicitement le refroidissement.
Ce que cela implique pour la découverte de matériaux
En apprenant à avancer les films atomiques par des sauts plus grands et physiquement fidèles, TrajCast peut générer des nanosecondes de données de trajectoire pour des systèmes contenant des milliers d’atomes dans le temps qu’il faudrait normalement pour simuler bien moins. La méthode conserve suffisamment de détails pour récupérer les observables clés, gère à la fois des molécules simples et des phases condensées complexes, et peut même extrapoler vers de nouveaux régimes comme la formation du verre sans entraînement supplémentaire. Bien que la version actuelle ne puisse pas encore prédire des propriétés dépendant de la pression et utilise un pas de prévision fixe, elle ouvre déjà la voie à un avenir où les simulations assistées par IA comblent le fossé entre les mouvements microscopiques rapides et les processus lents qui façonnent les matériaux et les liquides dans le monde réel.
Citation: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Mots-clés: dynamiques moléculaires, apprentissage automatique, simulation atomistique, science des matériaux, eau vitreuse