Clear Sky Science · pt
Dinâmica molecular sem forças por meio de redes autorregressivas equivariante
Por que acelerar os filmes atômicos importa
Muitas das questões mais instigantes na química, física e ciência dos materiais se resumem a como os átomos se movem: como uma molécula analgésica se contorce, como o quartzo vibra, como a água congela em formas vítreas estranhas. Experimentos computacionais chamados dinâmica molecular simulam esses filmes atômicos, mas são tão exigentes computacionalmente que os cientistas frequentemente veem apenas uma fração ínfima da ação. Este artigo apresenta o TrajCast, um novo arcabouço de inteligência artificial que aprende a avançar esses filmes atômicos sem sacrificar a física essencial.
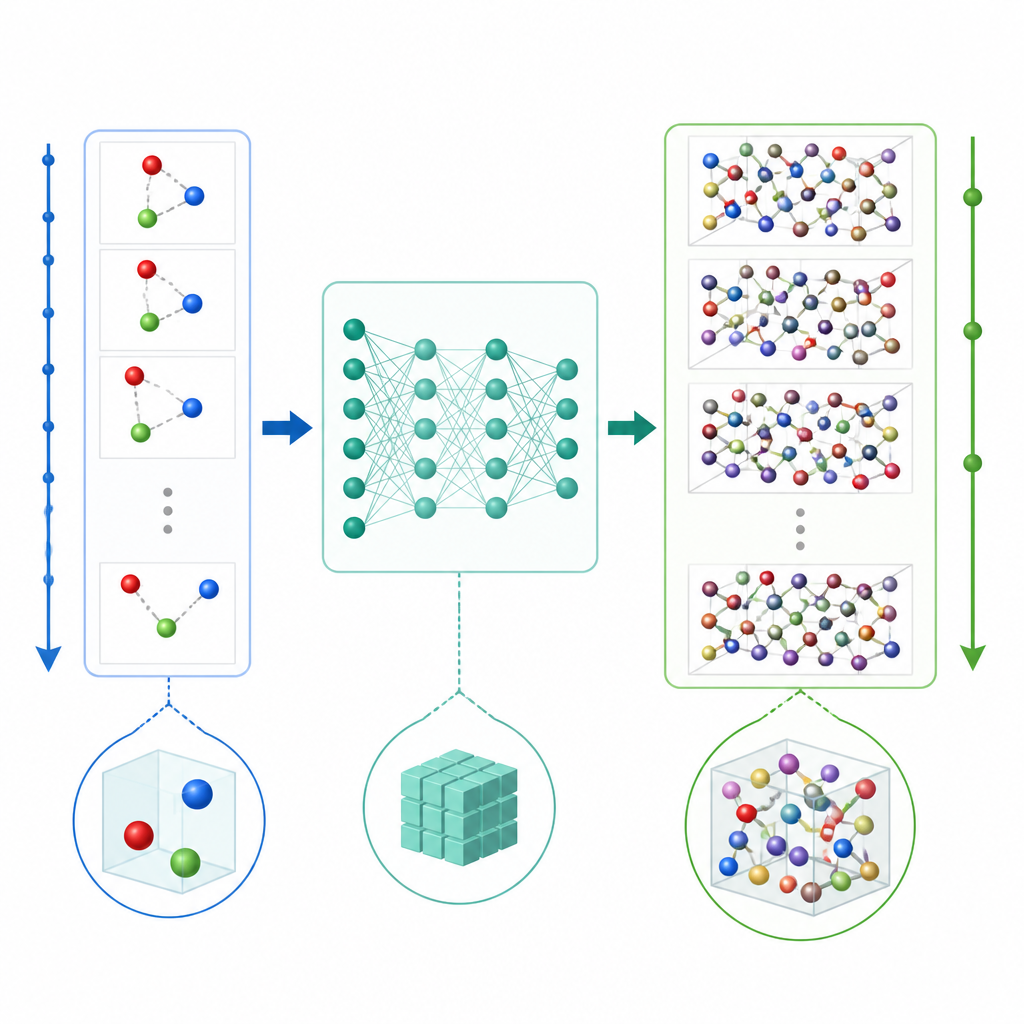
Um novo atalho para simular átomos
A dinâmica molecular tradicional atualiza o movimento atômico calculando repetidamente as forças entre átomos e então empurrando as posições em passos de tempo muito pequenos, frequentemente uma fração de um trilionésimo de segundo. Mesmo quando o aprendizado de máquina é usado para acelerar os cálculos de força, o passo de tempo diminuto ainda torna filmes longos caros. O TrajCast segue uma rota diferente. Em vez de prever forças, ele aprende a saltar diretamente de um instantâneo completo do sistema para o próximo, prevendo tanto onde cada átomo estará quanto quão rápido estará se movendo após um intervalo de tempo muito maior. Por baixo do capô, o TrajCast usa uma classe de redes neurais que respeitam as simetrias do espaço tridimensional para que rotacionar ou transladar o sistema não confunda o modelo.
Ensinar a IA a respeitar leis físicas
Para atuar como um substituto confiável da física, o TrajCast precisa fazer mais do que desenhar imagens plausíveis; deve reproduzir as estatísticas do movimento atômico real. Os autores treinam o modelo em simulações curtas e precisas que conservam energia e momento, e incorporam essas leis de conservação diretamente na rede para que os movimentos previstos permaneçam consistentes fisicamente. Durante longos “roll‑outs”, o TrajCast reutiliza repetidamente sua própria saída como a próxima entrada, enquanto um módulo termostato ajusta suavemente as velocidades atômicas para manter a temperatura simulada sob controle, assim como em simulações padrão de temperatura constante. Esse projeto permite ao método gerar trajetórias completas das quais os cientistas podem extrair propriedades dinâmicas, estruturais e energéticas.
Testando o TrajCast
A equipe avalia o TrajCast em três sistemas muito diferentes: uma única molécula de paracetamol na fase gasosa, quartzo cristalino e água líquida em condições ambiente. Para cada caso, comparam as trajetórias geradas pela IA com simulações convencionais, focando em medidas como como os átomos vibram, como a energia é distribuída e como as moléculas se movem ou difundem. No paracetamol, o TrajCast usa passos de tempo quatorze vezes maiores que a simulação de referência, e ainda reproduz de perto o espectro de movimentos vibracionais, a dispersão das energias potenciais e o panorama de barreiras energéticas que governam como a molécula alterna entre formas. No quartzo, ele pode tomar saltos com segurança trinta vezes maiores que o habitual, mantendo a correspondência com as assinaturas vibracionais e as flutuações de energia do sólido ao longo de nanossegundos.
Da água líquida a estados vítreos
A água líquida representa um desafio mais difícil porque as moléculas formam e quebram ligações de hidrogênio constantemente. Mesmo aqui, com um passo de tempo dez vezes maior, o TrajCast espelha as simulações de referência: captura modos vibracionais em várias frequências, reproduz a distância percorrida pelas moléculas de água ao longo do tempo e produz funções de distribuição radial que descrevem como as moléculas se organizam umas em relação às outras. Os autores então forçam o modelo bem além de sua zona de conforto de treinamento. Usando uma versão treinada apenas em água líquida à temperatura ambiente, eles resfriam gradualmente uma amostra maior para formar água vítrea hiperresfriada, um sólido desordenado com assinaturas estruturais sutis. Notavelmente, a IA acompanha como a difusão desacelera, como os picos estruturais se afinam e como medidas de ordem de curto e longo alcance evoluem, em estreita concordância com simulações dedicadas que incluem explicitamente o resfriamento.
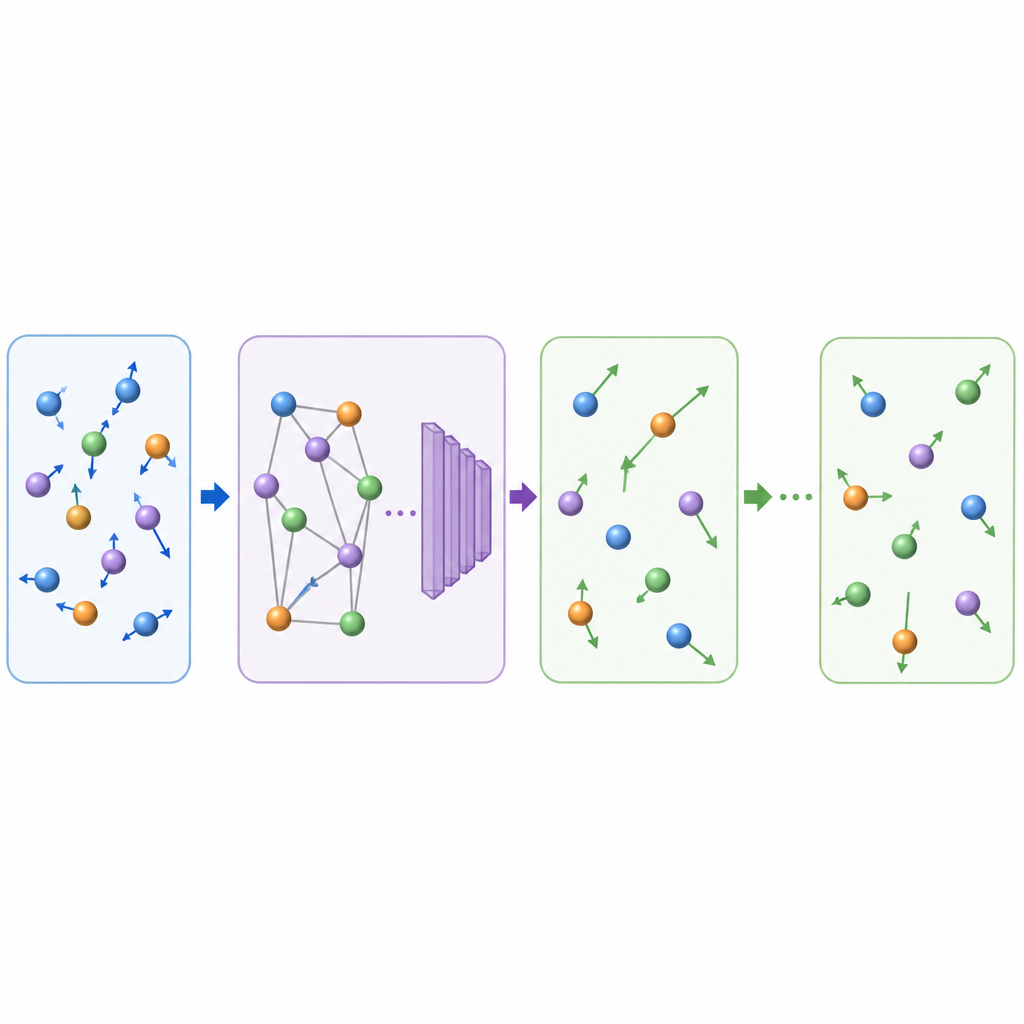
O que isso significa para a descoberta de materiais
Ao aprender a avançar filmes atômicos em saltos maiores e fisicamente fiéis, o TrajCast pode gerar nanossegundos de dados de trajetória para sistemas contendo milhares de átomos no tempo que normalmente levaria para simular muito menos. O método mantém detalhes suficientes para recuperar observáveis-chave, lida tanto com moléculas simples quanto com fases condensadas complexas, e pode até extrapolar para novos regimes, como formação de vidro, sem treinamento adicional. Embora a versão atual ainda não consiga prever propriedades dependentes da pressão e use um passo de previsão fixo, ela já aponta para um futuro no qual simulações assistidas por IA preenchem a lacuna entre movimentos microscópicos rápidos e os processos lentos que moldam materiais e líquidos no mundo real.
Citação: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Palavras-chave: dinâmica molecular, aprendizado de máquina, simulação atomística, ciência dos materiais, água vítrea