Clear Sky Science · sv
Force-free molecular dynamics through autoregressive equivariant networks
Varför det spelar roll att snabba upp atomfilmer
Många av de mest fascinerande frågorna inom kemi, fysik och materialvetenskap handlar om hur atomer rör sig: hur ett värkmedicinmolekyl vickar, hur kvartsvibrationer låter, hur vatten fryser till märkliga glaslika former. Datorexperiment kallade molekylär dynamik simulerar dessa atomfilmer, men de är så beräkningsintensiva att forskare ofta bara ser en mycket liten del av förloppet. Denna artikel presenterar TrajCast, en ny artificiell intelligens‑ramverk som lär sig att spola fram dessa atomfilmer utan att offra den väsentliga fysiken.
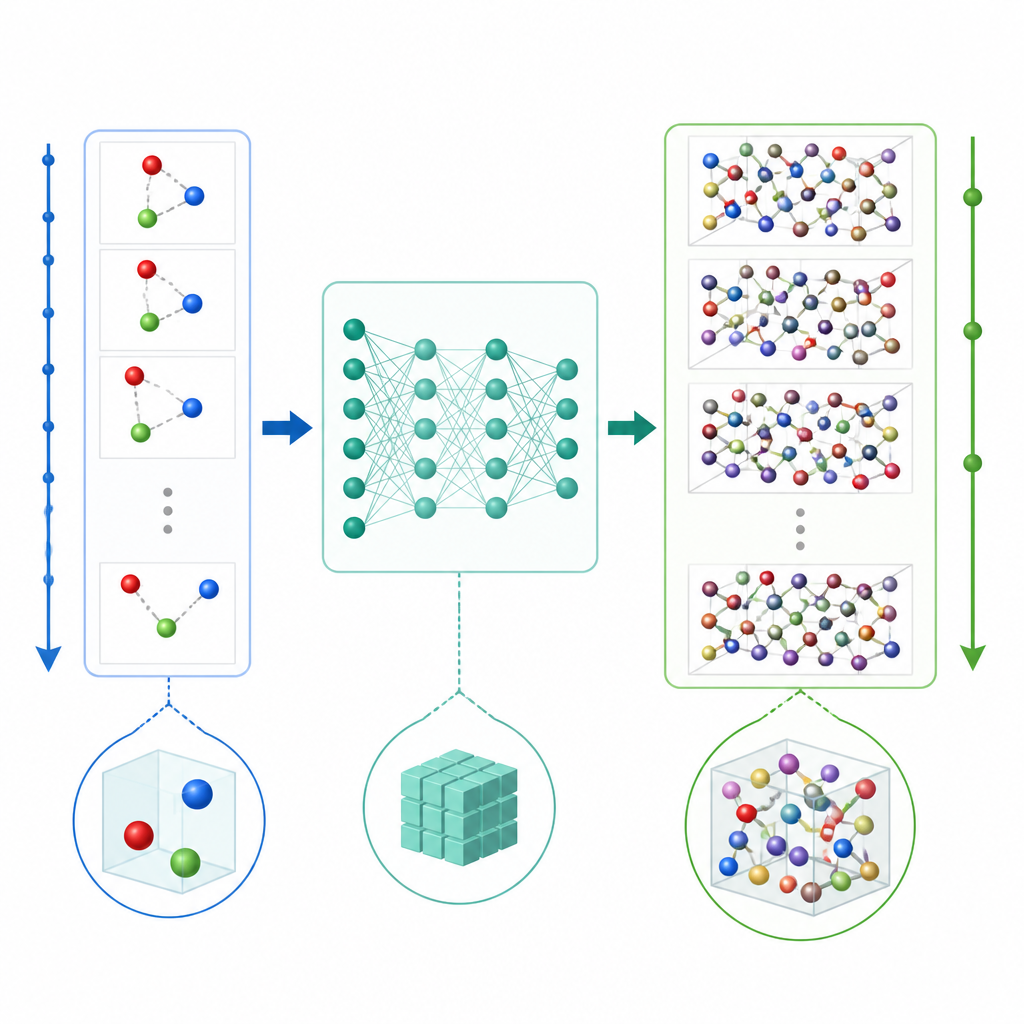
En ny genväg för att simulera atomer
Traditionell molekylär dynamik uppdaterar atomernas rörelse genom att upprepade gånger beräkna krafterna mellan atomerna och sedan skjuta fram positionerna i mycket små tidssteg, ofta en bråkdel av en biljonedel av en sekund. Även när maskininlärning används för att påskynda kraftberäkningarna gör det lilla tidssteget att långa filmer fortfarande blir dyra. TrajCast tar en annan väg. Istället för att förutsäga krafter lär den sig att hoppa direkt från en fullständig ögonblicksbild av systemet till nästa, och prognostiserar både var varje atom kommer att befinna sig och hur snabbt den kommer att röra sig efter ett mycket större tidsintervall. Under ytan använder TrajCast en klass av neurala nätverk som respekterar symmetrierna i tredimensionellt rum så att rotation eller translation av systemet inte förvirrar modellen.
Att lära AI att respektera fysikaliska lagar
För att fungera som en trovärdig ersättning för fysiken måste TrajCast göra mer än att rita trovärdiga bilder; det måste reproducera statistiken för verklig atomär rörelse. Författarna tränar modellen på korta, noggranna simuleringar som bevarar energi och rörelsemängd, och de bygger in dessa bevarandelagar direkt i nätverket så att de förutsagda rörelserna förblir fysiskt konsistenta. Under långa ”roll‑outs” använder TrajCast upprepade gånger sin egen utdata som nästa indata, medan en termostatmodul varsamt justerar atomernas hastigheter för att hålla den simulerade temperaturen under kontroll, precis som i standardiserade simuleringar vid konstant temperatur. Denna utformning gör att metoden kan generera fullständiga banor från vilka forskare kan extrahera dynamiska, strukturella och energimässiga egenskaper.
Sätta TrajCast på prov
Teamet jämför TrajCast mot tre mycket olika system: en enda paracetamolmolekyl i gasfas, kristallin kvarts och flytande vatten under rumsvillkor. För varje fall jämför de AI‑genererade banor med konventionella simuleringar, med fokus på mått som hur atomer vibrerar, hur energi fördelas och hur molekyler rör sig eller diffunderar. För paracetamol använder TrajCast tidssteg fjorton gånger större än referenssimuleringen, och ändå återger den väl spektrumet av vibrationsrörelser, spridningen av potentiella energier och energibarriärlandskapet som styr hur molekylen växlar mellan former. I kvarts kan den säkert ta hopp trettio gånger större än vanligt, samtidigt som den matchar de vibrerande signaturerna och energifluktuationerna i det fasta ämnet över nanosekunder.
Från flytande vatten till glaslika tillstånd
Flytande vatten utgör en tuffare utmaning eftersom molekyler ständigt bildar och bryter vätebindningar. Även här, med ett tio gånger större tidssteg, speglar TrajCast referenssimuleringarna: den fångar vibrationslägen över frekvenser, reproducerar hur långt vattenmolekyler rör sig över tid och ger radialfördelningsfunktioner som beskriver hur molekyler är ordnade gentemot varandra. Författarna pressar sedan modellen långt bortom dess träningskomfortzon. Med en version som endast tränats på rumstempererat flytande vatten kyler de gradvis ett större prov för att bilda hyperquenchad glasliknande vatten, ett oordnat fast ämne med subtila strukturella signaturer. Anmärkningsvärt nog följer AI:n hur diffusionen saktar in, hur strukturtopparna skärps och hur mått på långt‑ och kortsiktsordning utvecklas, i nära överensstämmelse med dedikerade simuleringar som explicit inkluderar avkylning.
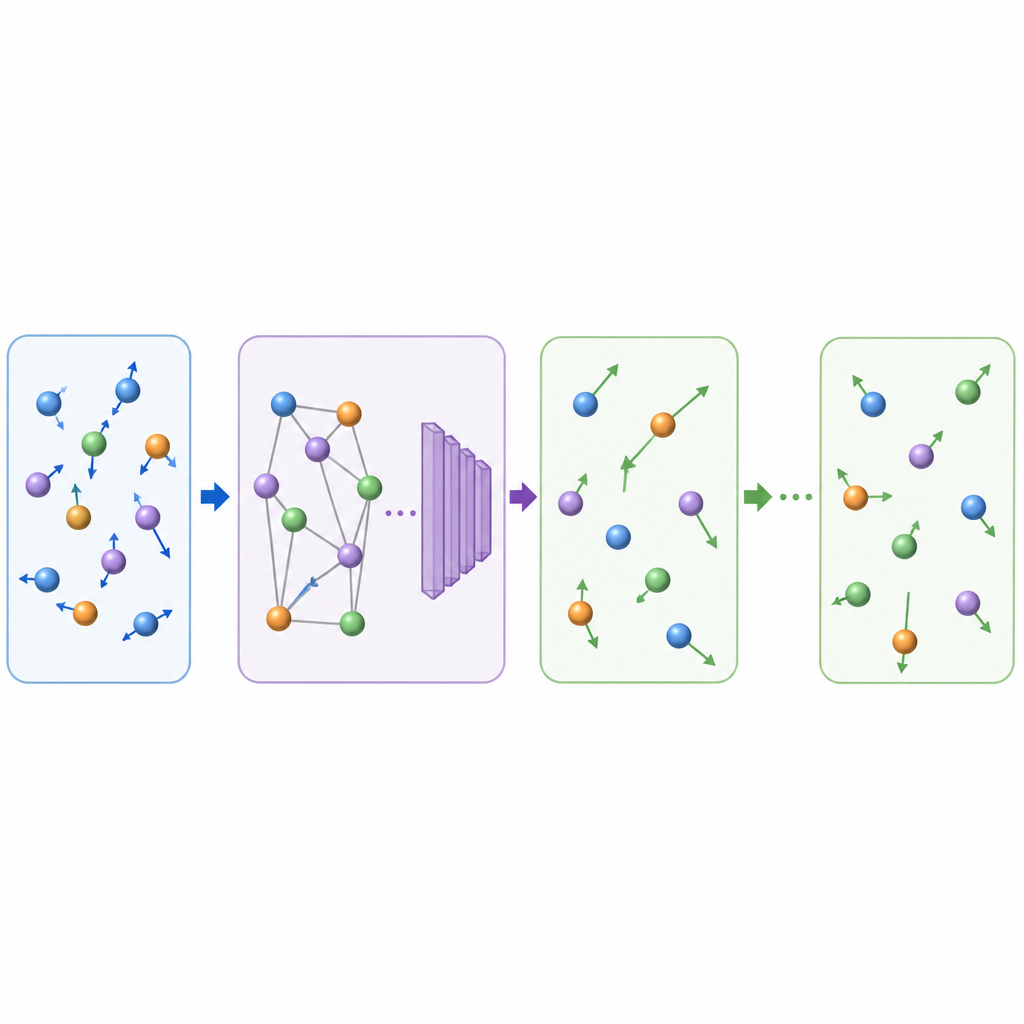
Vad detta betyder för framtida materialupptäckt
Genom att lära sig att avancera atomfilmer i större, fysikaliskt trogna hopp kan TrajCast generera nanosekunder av banafdata för system som innehåller tusentals atomer på den tid det normalt skulle ta att simulera betydligt mindre. Metoden behåller tillräckligt med detalj för att återfå nyckelobservationer, hanterar både enkla molekyler och komplexa kondenserade faser, och kan till och med extrapolera till nya regimer såsom glasbildning utan ytterligare träning. Även om den nuvarande versionen ännu inte kan förutsäga tryckberoende egenskaper och använder ett fast prognossteg, pekar den redan mot en framtid där AI‑stödja simuleringar bygger en bro mellan snabba mikroskopiska rörelser och de långsamma processer som formar material och vätskor i verkligheten.
Citering: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Nyckelord: molekylär dynamik, maskininlärning, atomistisk simulering, materialvetenskap, glassy water