Clear Sky Science · pl
Bezwładnościowa dynamika molekularna dzięki autoregresyjnym sieciom ekwiwariancyjnym
Dlaczego przyspieszanie „filmów” atomowych ma znaczenie
Wiele z najbardziej intrygujących zagadnień w chemii, fizyce i nauce o materiałach sprowadza się do tego, jak poruszają się atomy: jak porusza się cząsteczka środka przeciwbólowego, jak drga kwarc, jak woda zamarza, tworząc dziwne, szkliste formy. Eksperymenty komputerowe zwane dynamiką molekularną symulują te atomowe filmy, lecz są one tak wymagające obliczeniowo, że naukowcy często widzą tylko maleńki ułamek akcji. W artykule przedstawiono TrajCast, nową ramę sztucznej inteligencji, która uczy się przewijać te atomowe nagrania do przodu bez utraty istotnej fizyki.
Nowy skrót do symulowania atomów
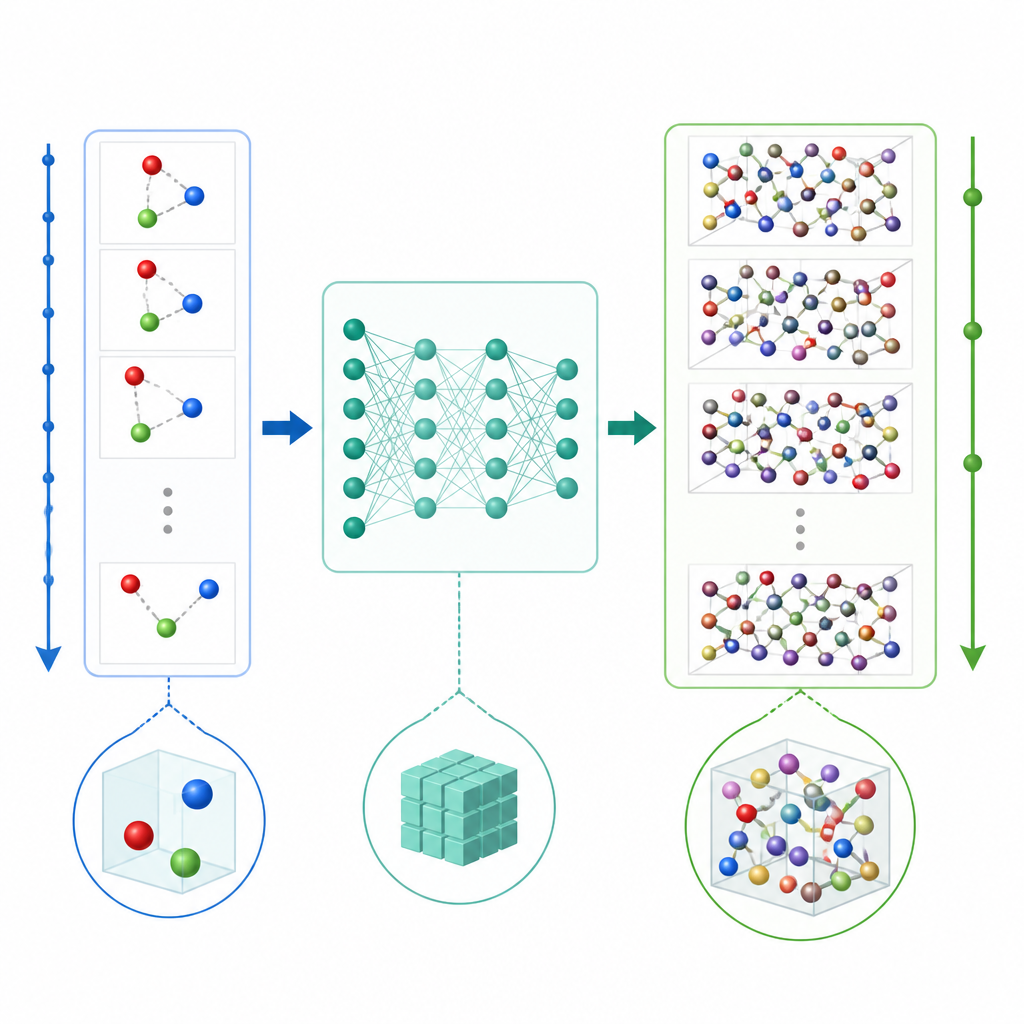
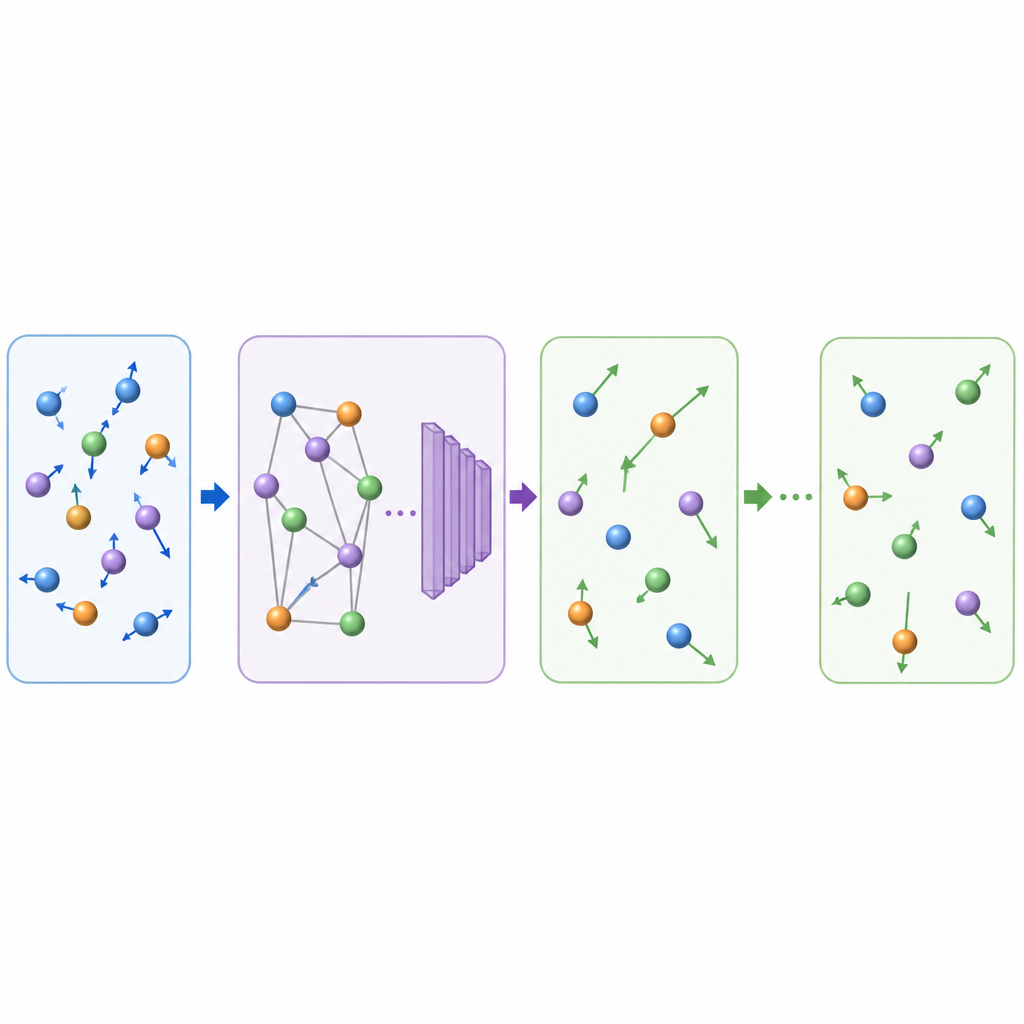
Tradycyjna dynamika molekularna aktualizuje ruch atomów przez wielokrotne obliczanie sił między atomami, a następnie przestawianie pozycji o bardzo małe kroki czasowe, często ułamki biliardowej części sekundy. Nawet gdy uczenie maszynowe przyspiesza obliczenia sił, drobny krok czasowy wciąż sprawia, że długie filmy są kosztowne. TrajCast podchodzi do problemu inaczej. Zamiast przewidywać siły, uczy się skakać bezpośrednio z jednej pełnej migawki układu do następnej, prognozując zarówno położenie każdego atomu, jak i jego prędkość po znacznie dłuższym czasie. W rdzeniu TrajCast wykorzystuje klasę sieci neuronowych, które respektują symetrie przestrzeni trójwymiarowej, tak aby rotacja czy translacja układu nie myliła modelu.
Nauka AI poszanowania zasad fizycznych
Aby pełnić wiarygodną rolę zastępcy fizyki, TrajCast musi robić więcej niż szkicować wiarygodne obrazy; musi odtwarzać statystyki rzeczywistego ruchu atomów. Autorzy trenują model na krótkich, dokładnych symulacjach, które zachowują energię i pęd, i wbudowują te prawa zachowania bezpośrednio w sieć, tak by przewidywane ruchy pozostały zgodne z fizyką. Podczas długich „rozwinąć” TrajCast wielokrotnie bierze własne wyjście jako następne wejście, a moduł termostatu delikatnie koryguje prędkości atomów, aby utrzymać kontrolę nad symulowaną temperaturą, podobnie jak w standardowych symulacjach w stałej temperaturze. Taka konstrukcja pozwala metodzie generować pełne trajektorie, z których naukowcy mogą wydobywać własności dynamiczne, strukturalne i energetyczne.
Testowanie TrajCast
Zespół sprawdza TrajCast na trzech bardzo różnych układach: jednej cząsteczce paracetamolu w fazie gazowej, krystalicznym kwarcu i ciekłej wodzie w warunkach pokojowych. W każdym przypadku porównują trajektorie generowane przez AI z konwencjonalnymi symulacjami, skupiając się na miarach takich jak drgania atomów, rozkład energii i ruch czy dyfuzja cząsteczek. Dla paracetamolu TrajCast używa kroków czasowych czternastokrotnie większych niż w symulacji odniesienia, a mimo to wiernie odtwarza spektrum drgań, rozkład energii potencjalnej i krajobraz barier energetycznych rządzących przejściami między kształtami cząsteczki. W kwarcu potrafi bezpiecznie wykonywać skoki trzydziestokrotnie większe niż zwykle, nadal dopasowując sygnatury drgań i fluktuacje energii ciała stałego na skali nanosekund.
Od ciekłej wody do stanów szklistych
Ciekła woda stanowi trudniejsze wyzwanie, ponieważ cząsteczki nieustannie tworzą i zrywają wiązania wodorowe. Nawet tutaj, przy dziesięciokrotnie większym kroku czasowym, TrajCast odzwierciedla symulacje referencyjne: uchwyca tryby drgań w różnych częstotliwościach, odwzorowuje, jak daleko przemieszczają się cząsteczki w czasie, i dostarcza radialnych funkcji rozkładu opisujących uporządkowanie molekuł względem siebie. Autorzy następnie pchają model daleko poza jego strefę komfortu treningowego. Używając wersji wytrenowanej jedynie na ciekłej wodzie w temperaturze pokojowej, stopniowo schładzają większą próbkę, aby utworzyć hiperquenched szklistą wodę — nieuporządkowane ciało stałe z subtelnymi sygnaturami strukturalnymi. Co niezwykłe, AI śledzi spowolnienie dyfuzji, wyostrzanie się piku strukturalnego i ewolucję miar porządku dalekiego i bliskiego zasięgu, w ścisłej zgodności z dedykowanymi symulacjami, które jawnie uwzględniają chłodzenie.
Co to oznacza dla przyszłych odkryć materiałowych
Ucząc się przesuwać „filmy” atomowe w większych, fizycznie wiernych skokach, TrajCast może wygenerować nanosekundy danych trajektorii dla układów zawierających tysiące atomów w czasie, który normalnie pozwoliłby zasymulować znacznie mniej. Metoda zachowuje wystarczającą szczegółowość, by odzyskać kluczowe obserwable, radzi sobie zarówno z prostymi cząsteczkami, jak i złożonymi fazami skondensowanymi, a nawet potrafi ekstrapolować do nowych reżimów, takich jak tworzenie szkła, bez dodatkowego treningu. Chociaż obecna wersja nie potrafi jeszcze przewidywać właściwości zależnych od ciśnienia i używa stałego kroku prognostycznego, już wskazuje na przyszłość, w której symulacje wspomagane przez AI zacierają przepaść między szybkimi mikroskopowymi ruchami a powolnymi procesami kształtującymi materiały i ciecze w rzeczywistym świecie.
Cytowanie: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Słowa kluczowe: dynamika molekularna, uczenie maszynowe, symulacja atomistyczna, nauka o materiałach, szklista woda