Clear Sky Science · de
Kraftfreie Molekulardynamik durch autoregressive äquivariante Netze
Warum es wichtig ist, atomare Filme zu beschleunigen
Viele der spannendsten Fragen in Chemie, Physik und Materialwissenschaft drehen sich um die Bewegung von Atomen: wie ein Schmerzmittelmolekül zappelt, wie Quarz vibriert oder wie Wasser in seltsame glasartige Formen gefriert. Computerversuche, sogenannte Molekulardynamik‑Simulationen, erzeugen diese atomaren Filme, sind aber so rechenintensiv, dass Forschende oft nur einen winzigen Ausschnitt des Geschehens sehen. Dieser Artikel stellt TrajCast vor, ein neues KI‑Framework, das lernt, diese atomaren Aufnahmen vorzuspulen, ohne die wesentliche Physik zu opfern.
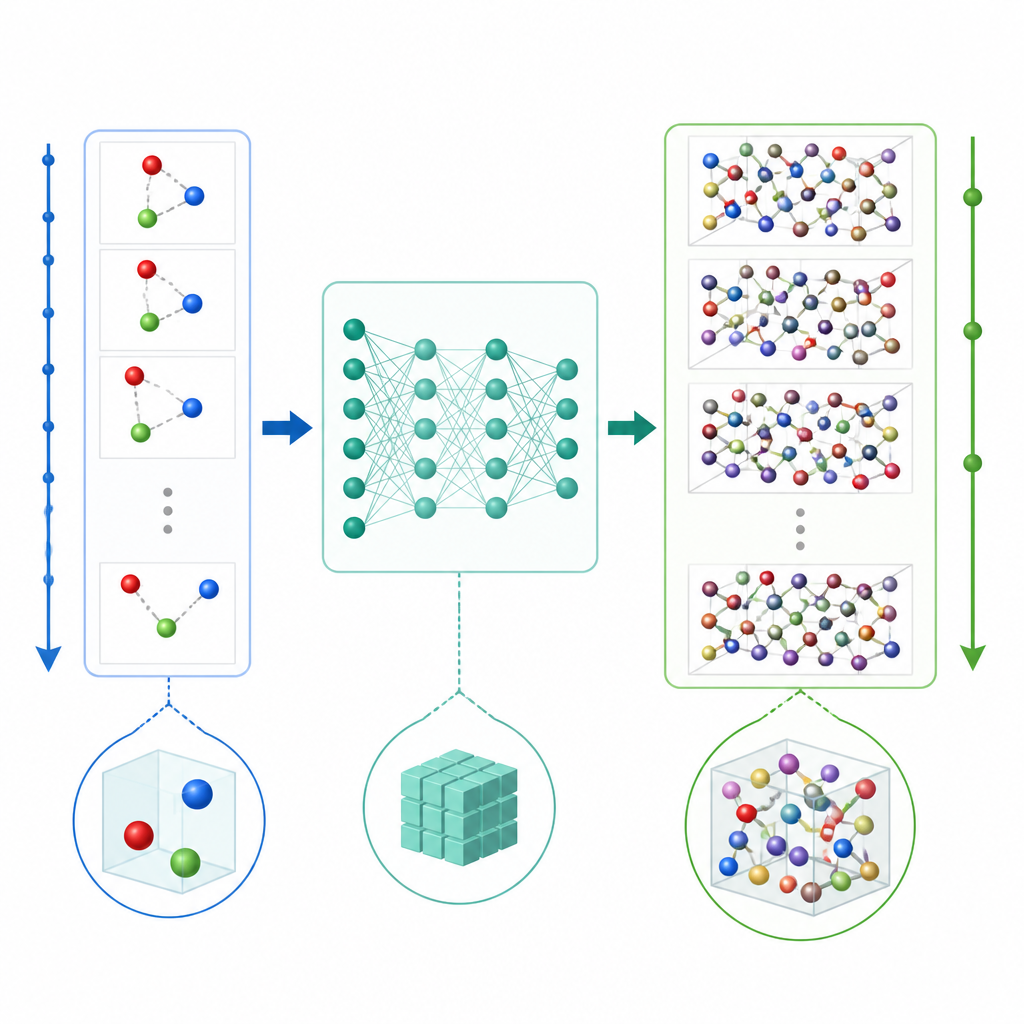
Eine neue Abkürzung zur Simulation von Atomen
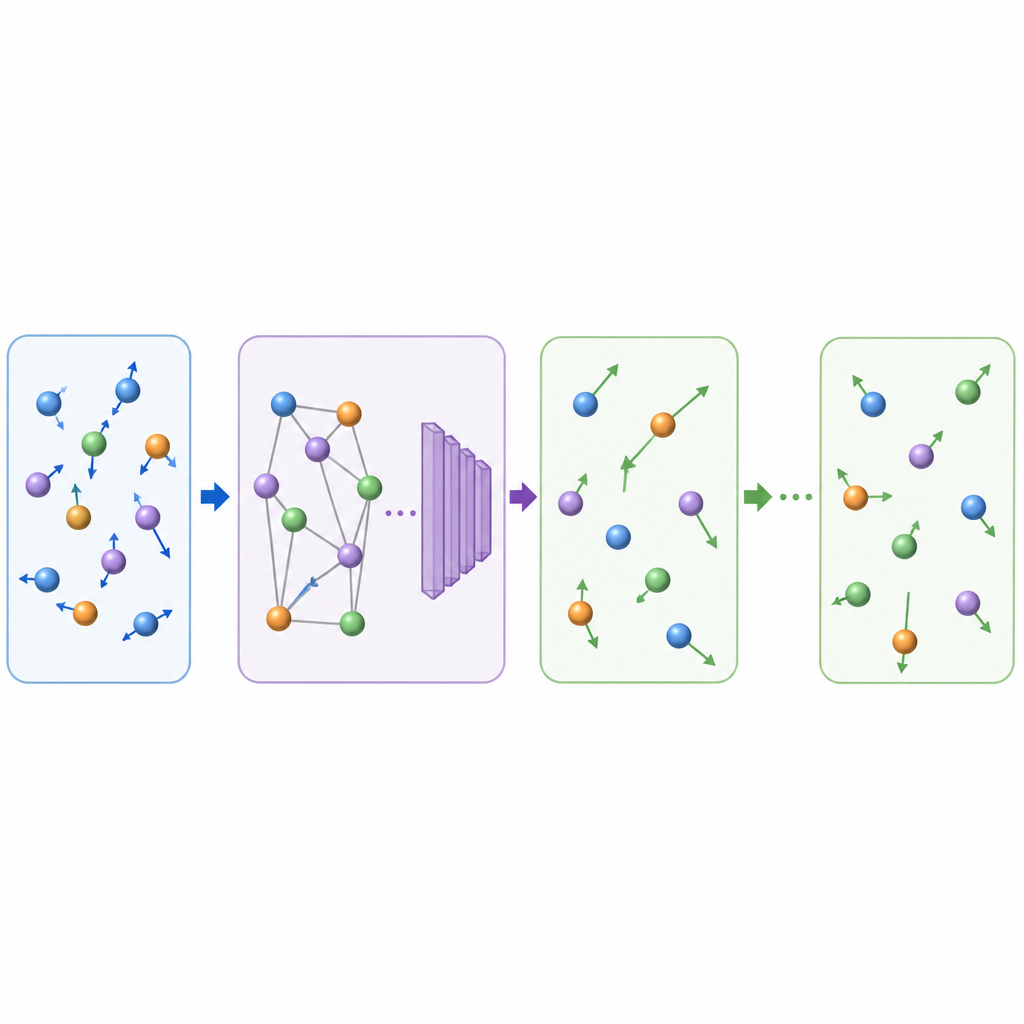
Traditionelle Molekulardynamik aktualisiert atomare Bewegungen, indem wiederholt die Kräfte zwischen Atomen berechnet und die Positionen in sehr kleinen Zeitschritten vorgerückt werden, oft ein Bruchteil einer Billionstel Sekunde. Selbst wenn maschinelles Lernen die Kraftberechnungen beschleunigt, bleibt der winzige Zeitschritt langwierige Filme teuer. TrajCast schlägt einen anderen Weg ein. Statt Kräfte vorherzusagen, lernt es, direkt von einer vollständigen Momentaufnahme des Systems zur nächsten zu springen und sowohl vorherzusagen, wo sich jedes Atom befinden wird, als auch wie schnell es sich nach einem deutlich längeren Zeitintervall bewegt. Unter der Haube nutzt TrajCast eine Klasse von neuronalen Netzen, die die Symmetrien des dreidimensionalen Raums respektiert, sodass Drehungen oder Verschiebungen des Systems das Modell nicht verwirren.
Der KI physikalische Regeln beibringen
Damit TrajCast als vertrauenswürdiger Stellvertreter der Physik fungieren kann, muss es mehr leisten als nur plausible Bilder zu erzeugen; es muss die Statistik realer atomarer Bewegungen reproduzieren. Die Autorinnen und Autoren trainieren das Modell an kurzen, genauen Simulationen, die Energie und Impuls erhalten, und bauen diese Erhaltungsgesetze direkt ins Netzwerk ein, sodass die vorhergesagten Bewegungen physikalisch konsistent bleiben. Bei langen "Roll‑outs" nimmt TrajCast wiederholt seine eigenen Ausgaben als nächsten Eingang, während ein Thermostatmodul die atomaren Geschwindigkeiten sanft anpasst, um die simulierte Temperatur unter Kontrolle zu halten, ähnlich wie in üblichen Simulationen bei konstanter Temperatur. Dieses Design erlaubt es der Methode, vollständige Trajektorien zu erzeugen, aus denen Forschende dynamische, strukturelle und energetische Eigenschaften extrahieren können.
TrajCast auf die Probe stellen
Das Team testet TrajCast an drei sehr unterschiedlichen Systemen: einem einzelnen Paracetamol‑Molekül in der Gasphase, kristallinem Quarz und flüssigem Wasser bei Raumbedingungen. Für jeden Fall vergleichen sie die KI‑generierten Trajektorien mit konventionellen Simulationen und konzentrieren sich auf Größen wie Vibrationsspektren, Energiedistributionen und Molekülbewegungen bzw. Diffusion. Für Paracetamol verwendet TrajCast Zeitschritte, die vierzehnmal größer sind als in der Referenzsimulation, und reproduziert dennoch eng das Spektrum der Schwingungsbewegungen, die Verteilung der potentiellen Energien und die Landschaft energetischer Barrieren, die das Umschlagen des Moleküls zwischen Formen steuern. In Quarz kann es sicher Sprünge dreißigmal größer als üblich machen und gleichzeitig die Vibrationssignaturen und Energiefluktuationen des Festkörpers über Nanosekunden hinweg abbilden.
Von flüssigem Wasser zu glasigen Zuständen
Flüssiges Wasser stellt eine größere Herausforderung dar, weil Moleküle ständig Wasserstoffbrücken bilden und brechen. Selbst hier spiegelt TrajCast mit einem zehnfach größeren Zeitschritt die Referenzsimulationen wider: Es fängt Schwingungsmoden über Frequenzen hinweg ein, reproduziert, wie weit Wassermoleküle sich über die Zeit bewegen, und liefert radiale Verteilungsfunktionen, die beschreiben, wie Moleküle zueinander angeordnet sind. Die Autorinnen und Autoren treiben das Modell dann weit über seine Trainingskonfiguration hinaus. Mit einer nur auf flüssigem Wasser bei Raumtemperatur trainierten Version kühlen sie schrittweise eine größere Probe ab, um hypergequenched glasiges Wasser zu formen, einen ungeordneten Festkörper mit subtilen Strukturmerkmalen. Bemerkenswerterweise verfolgt die KI, wie sich die Diffusion verlangsamt, wie Strukturpeaks sich schärfen und wie sich Maße lang‑ und kurzreichweitiger Ordnung entwickeln — in enger Übereinstimmung mit speziellen Simulationen, die das Abkühlen explizit einschließen.
Was das für die Entdeckung neuer Materialien bedeutet
Indem TrajCast lernt, atomare Filme in größeren, physikalisch treuen Sprüngen voranzutreiben, kann es Nanosekunden an Trajektoriendaten für Systeme mit Tausenden von Atomen in der Zeit erzeugen, die normalerweise für deutlich weniger Simulationen nötig wäre. Die Methode behält genug Details bei, um wichtige Observablen wiederherzustellen, handhabt sowohl einfache Moleküle als auch komplexe kondensierte Phasen und kann sogar ohne zusätzliches Training in neue Bereiche extrapolieren, etwa die Glasbildung. Obwohl die aktuelle Version noch keine druckabhängigen Eigenschaften vorhersagen kann und einen festen Vorhersageschritt verwendet, deutet sie bereits auf eine Zukunft hin, in der KI‑unterstützte Simulationen die Lücke zwischen schnellen mikroskopischen Bewegungen und den langsamen Prozessen schließen, die Materialien und Flüssigkeiten in der realen Welt formen.
Zitation: Thiemann, F.L., Reschützegger, T., Esposito, M. et al. Force-free molecular dynamics through autoregressive equivariant networks. Nat Mach Intell 8, 764–776 (2026). https://doi.org/10.1038/s42256-026-01227-7
Schlüsselwörter: molekulardynamik, maschinelles lernen, atomistische simulation, materialwissenschaft, glasiges Wasser