Clear Sky Science · zh
用于准确分子性质预测的分层交互消息网络
更聪明的捷径以发现更好的药物
将一种新药推向市场既缓慢又昂贵,部分原因在于化学家必须在实验室中测试大量分子,以确定它们是否安全、有效并在体内表现良好。本研究提出了一种新的人工智能模型,称为 HimNet,旨在仅凭分子结构更准确地预测这些关键的类药性质,帮助科研人员将实验集中在最有前途的候选者上。
为何预测分子行为如此困难
每一种潜在药物都必须满足一项被称为 ADMET 的严格清单:吸收(Absorption)、分布(Distribution)、代谢(Metabolism)、排泄(Excretion)以及毒性(Toxicity)。传统的计算模型要么依赖人工制定的化学规则,要么依赖将分子视为简单原子与键网络的深度学习系统。这些较早的方法常常忽视分子不同部分以微妙、非线性方式协同作用的情况——例如,两个相距较远的环如何堆叠,或一侧的极性基团如何削弱另一侧疏水片段的影响。因此,当化学家尝试探索全新的化学空间时,对溶解度、毒性或血脑屏障穿透性的预测可能不可靠。
分层观察分子
HimNet 通过同时在多个层次上观察每个分子来解决这一问题。最细的层次跟踪单个原子及其之间的键。第二层将原子分组成具有化学意义的“基元”(motifs),例如芳香环或酸性基团,而第三个全局层表示整个分子。HimNet 使用分层交互消息传递机制在这些层之间来回传递信息——本质上是一种结构化的对话,其中原子与基元交流,基元彼此交流,所有层共同构建对分子行为的总体认知。与此并行,模型还分析标准的分子“指纹”(fingerprints),这是一种在药物化学中广泛使用的紧凑数字编码,并学习这些指纹在同一分子的不同描述之间哪些方面最为一致。
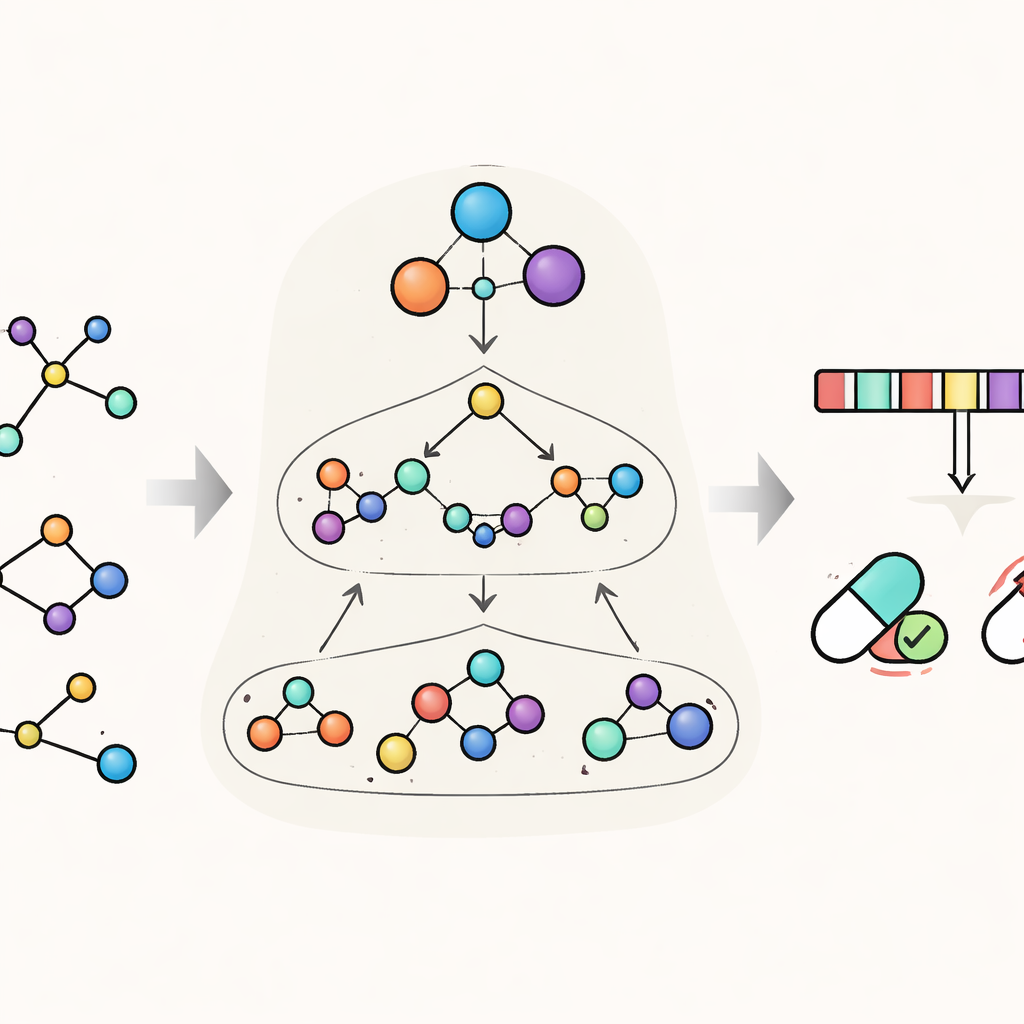
教会模型聚焦重要部分
HimNet 的核心思想之一是注意力:模型学习突出哪些原子和基元对某一特定性质最为重要,以及它们的影响如何相互增强或抵消。网络中的一条路径严格沿实际化学键展开,保留化学家所信赖的局部细节。另一条路径则在分子内部构建更远程的“桥梁”,允许相距较远的基团相互作用,即使它们被若干键分隔。一个可学习的门控机制将这两条信息流融合,平衡严格的化学连接性与灵活的全局上下文。额外的模块将分层图视图与指纹视图对齐,给出一个最终的紧凑表示,既捕捉细微结构又体现更广泛的化学模式。
将方法付诸考验
为了检验该设计的效果,作者在十一个数据集上评估了 HimNet,涵盖药物发现中许多重要任务。这些任务包括经典的毒性基准、血脑屏障穿透性以及诸如溶解度等基本物理属性,还有更具挑战性的真实世界问题,例如代谢稳定性、抗疟活性以及不同物种的肝脏清除率。在大多数任务中,HimNet 能够匹配或超越现有的最佳模型,且常常具有明显优势。重要的是,该模型在具有挑战性的数据集上仍然保持鲁棒,尽管在非常小、噪声高或类别高度不平衡的数据集中性能有所下降——这类情况对任何方法来说都很困难。消融研究(系统性地移除架构的部分)表明,从跨层注意力到指纹融合的每个组成模块都对准确性有可测的贡献。
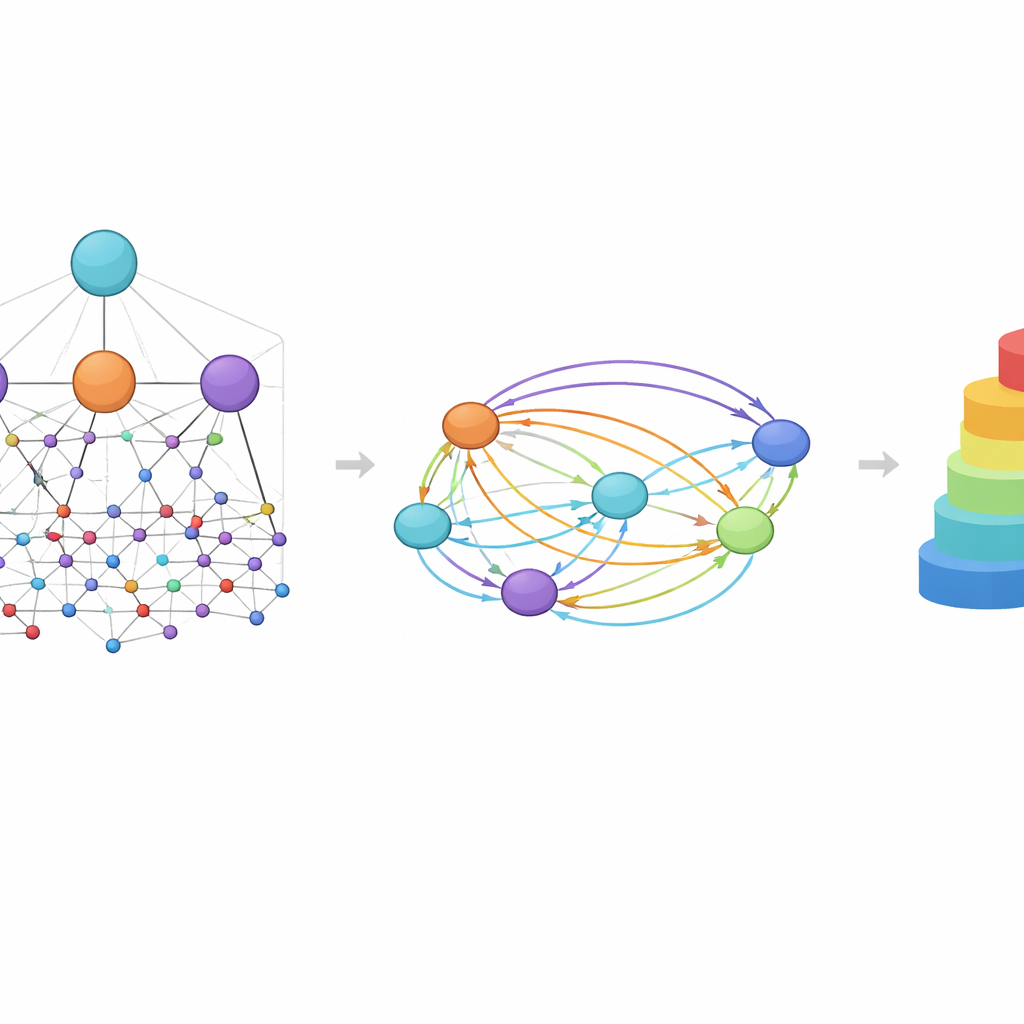
透过模型之眼看化学
由于 HimNet 在多个层次上使用注意力机制,其预测可以以化学家能解读的方式进行可视化。在关于血脑屏障渗透性的案例研究中,增加渗透性的分子区域以暖色显示,而阻碍渗透性的特征则以冷色显示。疏水性环和氟化基元往往提高穿透性,而高度极性的基团,例如羧酸或某些羰基和含氮中心,通常会降低它。引人注目的是,模型有时会根据环境以不同方式对待两个看似相似的环,这反映了真实化学语境如何将分子的某一部分变为有利或不利。
这对未来药物意味着什么
对非专业读者来说,关键信息是 HimNet 提供了一种更为细致的 AI“解读”分子的方法:分子不仅仅是原子列表,而是相互作用的部分,其组合行为决定了化合物是否可能成为良好的药物。通过捕捉这些多层交互并以化学上合理的方式解释其关注点,HimNet 可以帮助研究人员更有信心地筛选庞大的虚拟化合物库。尽管该方法计算开销更大,且仍需扩展以处理完整三维构型和非常柔性的分子,但它指向了更智能、更透明的工具,这些工具有望缩短从分子构想到安全有效药物的路径。
引用: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
关键词: 分子性质预测, 图神经网络, 药物发现, ADMET 建模, 可解释人工智能