Clear Sky Science · de
Ein hierarchisches Interaction-Message-Netz für präzise Vorhersagen molekularer Eigenschaften
Intelligentere Abkürzungen zur Entwicklung besserer Medikamente
Ein neues Medikament auf den Markt zu bringen ist langsam und teuer, unter anderem weil Chemiker sehr viele Moleküle im Labor testen müssen, um herauszufinden, ob sie sicher, wirksam und im Körper gut verträglich sind. Diese Studie stellt ein neues KI-Modell namens HimNet vor, das entwickelt wurde, um diese entscheidenden medikamentenähnlichen Eigenschaften deutlich genauer allein aus der Struktur eines Moleküls vorherzusagen und so Wissenschaftlern zu helfen, Experimente auf die vielversprechendsten Kandidaten zu konzentrieren.
Warum die Vorhersage des Verhaltens von Molekülen so schwierig ist
Jedes potenzielle Medikament muss eine anspruchsvolle Checkliste erfüllen, bekannt als ADMET: Absorption, Verteilung, Metabolismus, Ausscheidung und Toxizität. Traditionelle Computermodelle stützen sich entweder auf handgefertigte chemische Regeln oder auf Deep-Learning-Systeme, die Moleküle als einfache Netzwerke aus Atomen und Bindungen betrachten. Diese älteren Ansätze übersehen oft, wie verschiedene Teile eines Moleküls auf subtile, nicht-additive Weise zusammenwirken – zum Beispiel wie zwei weit auseinanderliegende Ringe übereinander stapeln können oder wie eine polare Gruppe auf einer Seite den Effekt eines fettigen Abschnitts auf der anderen Seite abschwächt. Infolgedessen können Vorhersagen zu Löslichkeit, Toxizität oder der Blut-Hirn-Schranken-Durchgängigkeit unzuverlässig sein, wenn Chemiker versuchen, neue Bereiche des chemischen Raums zu erkunden.
Moleküle in Schichten betrachten
HimNet geht dieses Problem an, indem es jedes Molekül gleichzeitig auf mehreren Ebenen betrachtet. Auf der feinsten Ebene verfolgt es einzelne Atome und die dazwischen liegenden Bindungen. Eine zweite Ebene gruppiert Atome zu chemisch sinnvollen „Motiven“, etwa aromatischen Ringen oder sauren Gruppen, während eine dritte, globale Ebene das ganze Molekül repräsentiert. HimNet lässt Informationen zwischen diesen Schichten über einen hierarchischen Interaction-Message-Passing-Mechanismus hin- und hersenden – im Wesentlichen ein strukturiertes Gespräch, bei dem Atome mit Motiven kommunizieren, Motive untereinander sprechen und alles zu einem Gesamtbild des molekularen Verhaltens beiträgt. Parallel dazu analysiert das Modell auch gängige molekulare „Fingerprints“, kompakte digitale Codierungen, die in der medizinischen Chemie weit verbreitet sind, und lernt, welche Aspekte dieser Fingerprints über verschiedene Beschreibungen desselben Moleküls hinweg am konsistentesten sind.
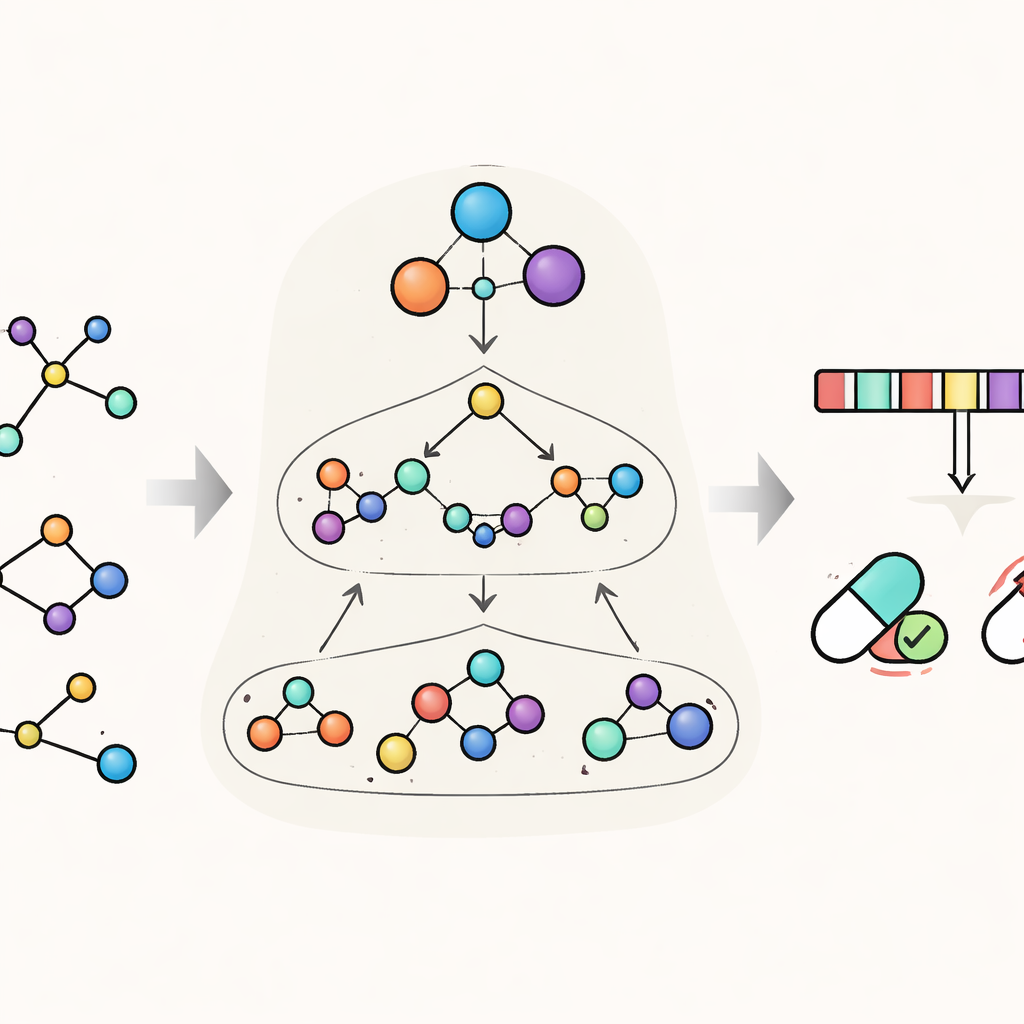
Dem Modell beibringen, sich auf das Wesentliche zu konzentrieren
Eine zentrale Idee in HimNet ist Attention: Das Modell lernt zu betonen, welche Atome und Motive für eine bestimmte Eigenschaft am wichtigsten sind und wie sich ihre Einflüsse gegenseitig verstärken oder ausgleichen. Ein Pfad im Netzwerk folgt sorgfältig den tatsächlichen chemischen Bindungen und bewahrt die lokalen Details, denen Chemiker vertrauen. Ein zweiter Pfad baut längerreichende „Brücken“ über das Molekül hinweg, sodass entfernte Gruppen miteinander interagieren können, selbst wenn sie durch mehrere Bindungen getrennt sind. Ein lernbares Gate mischt diese beiden Ströme und balanciert strikte chemische Konnektivität mit flexiblem, globalem Kontext. Zusätzliche Module bringen die geschichtete Graphansicht mit der Fingerprint-Ansicht in Einklang und liefern eine abschließende kompakte Repräsentation, die sowohl die feine Struktur als auch breitere chemische Muster erfasst.
Den Ansatz auf die Probe stellen
Um zu prüfen, wie gut dieses Design funktioniert, bewerteten die Autoren HimNet an elf Datensätzen, die viele Aufgaben abdecken, die in der Arzneimittelentwicklung wichtig sind. Dazu gehören klassische Benchmarks für Toxizität, Blut-Hirn-Schranken-Durchlässigkeit und grundlegende physikalische Eigenschaften wie Löslichkeit sowie anspruchsvollere, reale Probleme wie metabolische Stabilität, antimalarielle Aktivität und Leberclearance in verschiedenen Spezies. In den meisten dieser Aufgaben übertraf oder erreichte HimNet die besten bestehenden Modelle, oft mit deutlichem Abstand. Wichtig ist, dass das Modell auch bei schwierigen Datensätzen robust blieb, wobei die Leistung allerdings bei sehr kleinen, verrauschten oder stark unausgeglichenen Datensammlungen, mit denen jede Methode Probleme hat, etwas nachließ. Ablationsstudien – das systematische Entfernen von Architekturteilen – zeigten, dass jede Komponente, vom Cross-Level-Attention bis zur Fingerprint-Fusion, einen messbaren Beitrag zur Genauigkeit leistet.
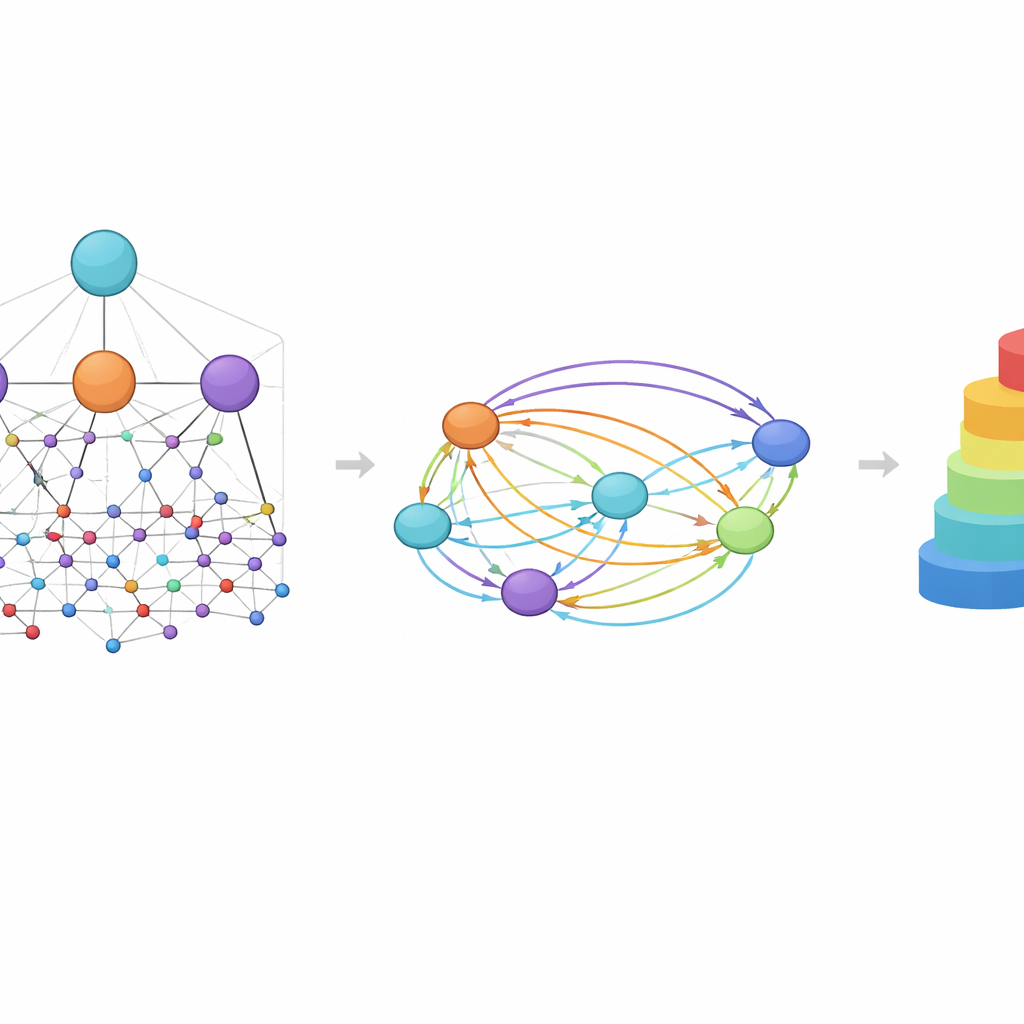
Chemie durch die Augen des Modells sehen
Weil HimNet Attention auf mehreren Ebenen einsetzt, lassen sich seine Vorhersagen so visualisieren, dass Chemiker sie interpretieren können. In Fallstudien zur Durchgängigkeit der Blut-Hirn-Schranke wurden Molekülregionen, die die Penetration erhöhten, in warmen Farben hervorgehoben, während Merkmale, die die Penetration behinderten, kühl dargestellt wurden. Hydrophobe Ringe und fluorierte Motive erhöhten tendenziell die Durchlässigkeit, während stark polare Gruppen, wie Carbonsäuren oder bestimmte Carbonyl- und Stickstoffzentren, sie oft verringerten. Auffällig behandelte das Modell manchmal zwei scheinbar ähnliche Ringe unterschiedlich, abhängig von ihrem Umfeld, was widerspiegelt, wie chemischer Kontext einen Teil eines Moleküls zu einem Vorteil und einen anderen zu einem Nachteil machen kann.
Was das für zukünftige Medikamente bedeutet
Für Nichtfachleute ist die Kernbotschaft, dass HimNet eine nuanciertere Möglichkeit bietet, wie KI Moleküle „liest“: nicht nur als Listen von Atomen, sondern als interagierende Teile, deren kombiniertes Verhalten bestimmt, ob eine Verbindung als gutes Medikament in Frage kommt. Indem es diese mehrschichtigen Interaktionen erfasst und seine Aufmerksamkeit in chemisch sinnvollen Begriffen erklärt, kann HimNet Forschern helfen, riesige virtuelle Bibliotheken mit größerer Zuversicht zu durchforsten. Obwohl die Methode rechenintensiver ist und noch Erweiterungen braucht, um vollständige dreidimensionale Gestalt und sehr flexible Moleküle zu berücksichtigen, zeigt sie den Weg zu intelligenteren, transparenteren Werkzeugen, die den Weg von der molekularen Idee zum sicheren und wirksamen Medikament verkürzen könnten.
Zitation: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Schlüsselwörter: Vorhersage molekularer Eigenschaften, Graph-Neuronale Netze, Arzneimittelentdeckung, ADMET-Modellierung, erklärbare KI