Clear Sky Science · en
A hierarchical interaction message net for accurate molecular property prediction
Smarter Shortcuts for Finding Better Medicines
Bringing a new medicine to market is slow and expensive, in part because chemists must test huge numbers of molecules in the lab to see whether they are safe, effective, and behave well in the body. This study introduces a new artificial intelligence model, called HimNet, designed to predict these crucial drug-like properties far more accurately from a molecule’s structure alone, helping scientists focus experiments on the most promising candidates.
Why Predicting Molecule Behavior Is So Hard
Every potential drug must satisfy a demanding checklist known as ADMET: how it is absorbed, distributed, metabolized, excreted, and whether it is toxic. Traditional computer models either rely on hand-crafted chemical rules or on deep-learning systems that see molecules as simple networks of atoms and bonds. These older approaches often miss how different parts of a molecule work together in subtle, non-additive ways—for example, how two distant rings might stack, or how a polar group on one side can weaken the effect of a greasy patch on the other. As a result, predictions about solubility, toxicity, or blood–brain barrier penetration can be unreliable when chemists try to explore new regions of chemical space.
Looking at Molecules in Layers
HimNet tackles this problem by viewing each molecule on several levels at once. At the finest level, it tracks individual atoms and the bonds between them. A second level groups atoms into chemically meaningful “motifs,” such as aromatic rings or acidic groups, while a third, global level represents the whole molecule. HimNet passes information back and forth across these layers using a hierarchical interaction message passing mechanism—essentially a structured conversation in which atoms talk to motifs, motifs talk to each other, and everything contributes to an overall picture of the molecule’s behavior. In parallel, the model also analyzes standard molecular “fingerprints,” compact digital encodings widely used in medicinal chemistry, and learns which aspects of these fingerprints are most consistent across different descriptions of the same molecule.
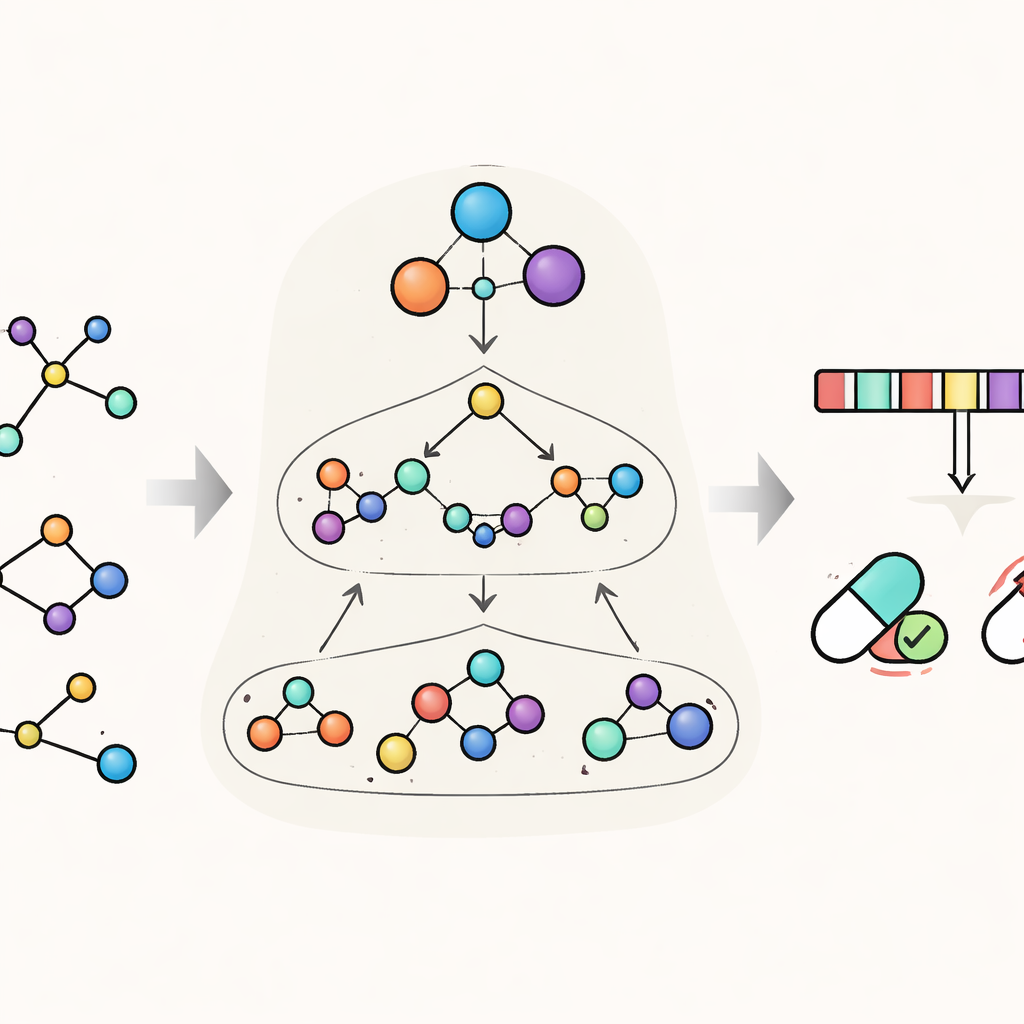
Teaching the Model to Focus on What Matters
A central idea in HimNet is attention: the model learns to highlight which atoms and motifs are most important for a given property and how their influences reinforce or cancel one another. One path in the network carefully follows the actual chemical bonds, preserving the local details that chemists trust. A second path builds longer-range “bridges” across the molecule, allowing distant groups to interact even if they are separated by several bonds. A learnable gate blends these two streams, balancing strict chemical connectivity with flexible, global context. Additional modules align the layered graph view with the fingerprint view, giving a final compact representation that captures both the fine structure and the broader chemical patterns.
Putting the Approach to the Test
To see how well this design works, the authors evaluated HimNet on eleven datasets covering many tasks that matter in drug discovery. These include classic benchmarks for toxicity, blood–brain barrier penetration, and basic physical properties like solubility, as well as more demanding, real-world problems such as metabolic stability, antimalarial activity, and liver clearance in different species. Across most of these tasks, HimNet matched or surpassed the best existing models, often with a clear margin. Importantly, the model remained robust even on challenging datasets, though performance dipped somewhat for very small, noisy, or highly imbalanced collections where any method struggles. Ablation studies—systematically removing parts of the architecture—showed that each component, from cross-level attention to fingerprint fusion, makes a measurable contribution to accuracy.
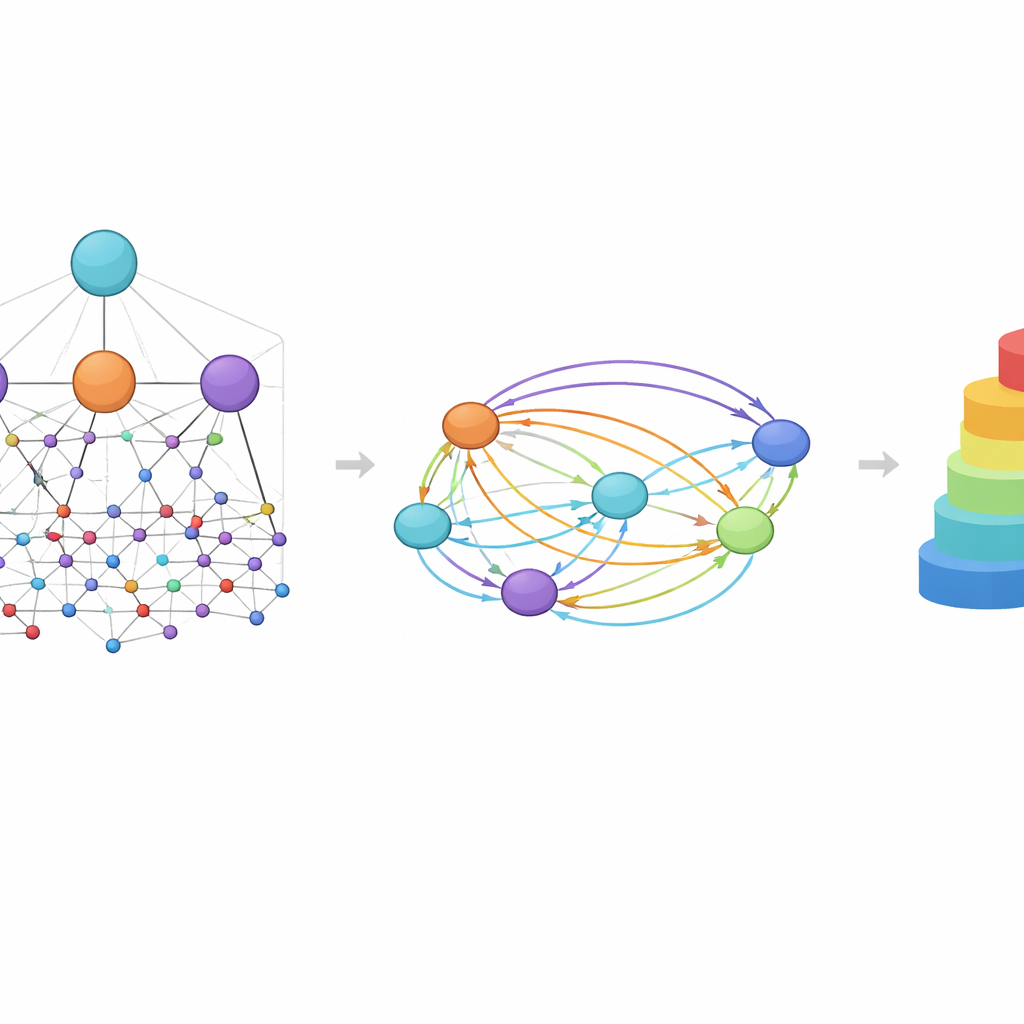
Seeing Chemistry Through the Model’s Eyes
Because HimNet uses attention at multiple levels, its predictions can be visualized in ways that chemists can interpret. In case studies on blood–brain barrier permeability, regions of a molecule that increase penetration were highlighted in warm colors, while features that hinder penetration appeared cool. Hydrophobic rings and fluorinated motifs tended to boost permeability, whereas highly polar groups, such as carboxylic acids or certain carbonyl and nitrogen centers, often reduced it. Strikingly, the model sometimes treated two seemingly similar rings differently depending on their surroundings, mirroring how real chemical context can turn one part of a molecule into a help and another into a hindrance.
What This Means for Future Medicines
For non-specialists, the key message is that HimNet offers a more nuanced way for AI to “read” molecules, not just as lists of atoms but as interacting parts whose combined behavior determines whether a compound is likely to make a good drug. By capturing these multi-layer interactions and explaining its focus in chemically sensible terms, HimNet can help researchers sift through vast virtual libraries with greater confidence. While the method is more computationally demanding and still needs extensions to handle full three-dimensional shape and very flexible molecules, it points toward smarter, more transparent tools that could shorten the path from molecular idea to safe and effective medicine.
Citation: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Keywords: molecular property prediction, graph neural networks, drug discovery, ADMET modeling, explainable AI