Clear Sky Science · es
Una red de mensajes de interacción jerárquica para la predicción precisa de propiedades moleculares
Atajos más inteligentes para encontrar mejores medicinas
Llevar un nuevo medicamento al mercado es lento y costoso, en parte porque los químicos deben ensayar en el laboratorio un número enorme de moléculas para comprobar si son seguras, eficaces y se comportan bien en el organismo. Este estudio presenta un nuevo modelo de inteligencia artificial, llamado HimNet, diseñado para predecir estas propiedades cruciales de tipo farmacológico con mucha más precisión a partir de la estructura de una molécula, ayudando a los científicos a centrar los experimentos en los candidatos más prometedores.
Por qué es tan difícil predecir el comportamiento de una molécula
Cada posible fármaco debe satisfacer una lista exigente conocida como ADMET: cómo se absorbe, distribuye, metaboliza, elimina y si es tóxico. Los modelos informáticos tradicionales se basan en reglas químicas manuales o en sistemas de aprendizaje profundo que ven las moléculas como redes simples de átomos y enlaces. Estos enfoques más antiguos a menudo pasan por alto cómo distintas partes de una molécula funcionan conjuntamente de maneras sutiles y no aditivas —por ejemplo, cómo dos anillos distantes pueden apilarse o cómo un grupo polar en un lado puede atenuar el efecto de una zona lipofílica en el otro—. Como resultado, las predicciones sobre la solubilidad, la toxicidad o la penetración de la barrera hematoencefálica pueden ser poco fiables cuando los químicos exploran nuevas regiones del espacio químico.
Mirar las moléculas en capas
HimNet aborda este problema viendo cada molécula en varios niveles a la vez. En el nivel más fino rastrea los átomos individuales y los enlaces entre ellos. Un segundo nivel agrupa átomos en “motivos” químicamente significativos, como anillos aromáticos o grupos ácidos, mientras que un tercer nivel global representa la molécula completa. HimNet hace circular información entre estas capas mediante un mecanismo jerárquico de paso de mensajes de interacción —esencialmente una conversación estructurada en la que los átomos hablan con los motivos, los motivos se comunican entre sí y todo contribuye a una imagen global del comportamiento de la molécula. En paralelo, el modelo también analiza las “huellas” moleculares estándar, codificaciones digitales compactas ampliamente usadas en química médica, y aprende qué aspectos de estas huellas son más coherentes entre distintas descripciones de la misma molécula.
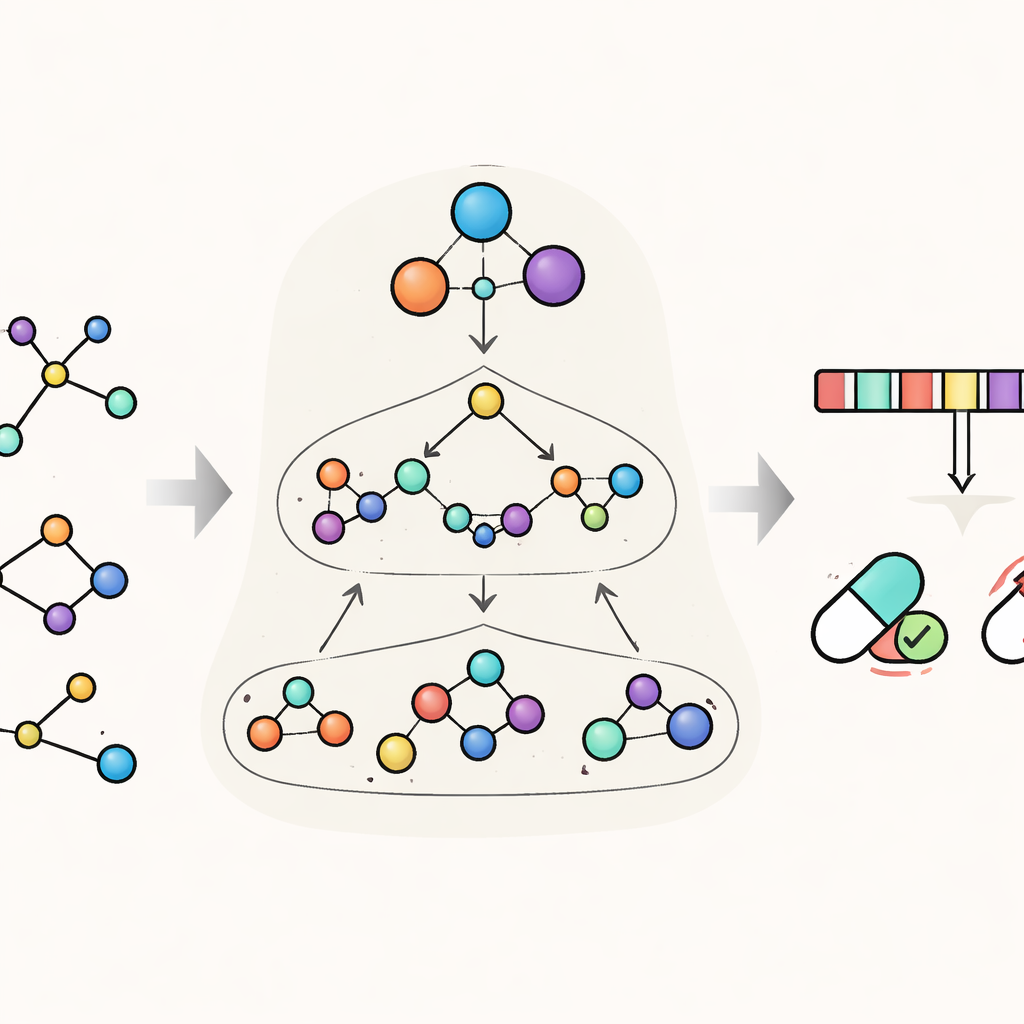
Enseñar al modelo a centrarse en lo que importa
Una idea central en HimNet es la atención: el modelo aprende a resaltar qué átomos y motivos son más importantes para una propiedad dada y cómo sus influencias se refuerzan o se cancelan entre sí. Un camino en la red sigue cuidadosamente los enlaces químicos reales, preservando los detalles locales en los que confían los químicos. Un segundo camino construye “puentes” de mayor alcance a lo largo de la molécula, permitiendo que grupos distantes interactúen incluso si están separados por varios enlaces. Una compuerta aprendible mezcla estas dos corrientes, equilibrando la conectividad química estricta con un contexto global más flexible. Módulos adicionales alinean la vista en capas del grafo con la vista de huellas, dando una representación final compacta que captura tanto la estructura fina como los patrones químicos más amplios.
Poner el enfoque a prueba
Para evaluar la eficacia de este diseño, los autores probaron HimNet en once conjuntos de datos que abarcan muchas tareas relevantes en el descubrimiento de fármacos. Estos incluyen puntos de referencia clásicos para toxicidad, penetración de la barrera hematoencefálica y propiedades físicas básicas como la solubilidad, así como problemas del mundo real más exigentes, como estabilidad metabólica, actividad antipalúdica y aclaramiento hepático en distintas especies. En la mayoría de estas tareas, HimNet igualó o superó a los mejores modelos existentes, a menudo con un margen claro. De manera importante, el modelo se mantuvo robusto incluso en conjuntos de datos desafiantes, aunque el rendimiento disminuyó algo en colecciones muy pequeñas, ruidosas o altamente desequilibradas, donde cualquier método tiene dificultades. Estudios de ablación —eliminando sistemáticamente partes de la arquitectura— mostraron que cada componente, desde la atención entre niveles hasta la fusión de huellas, aporta una contribución medible a la precisión.
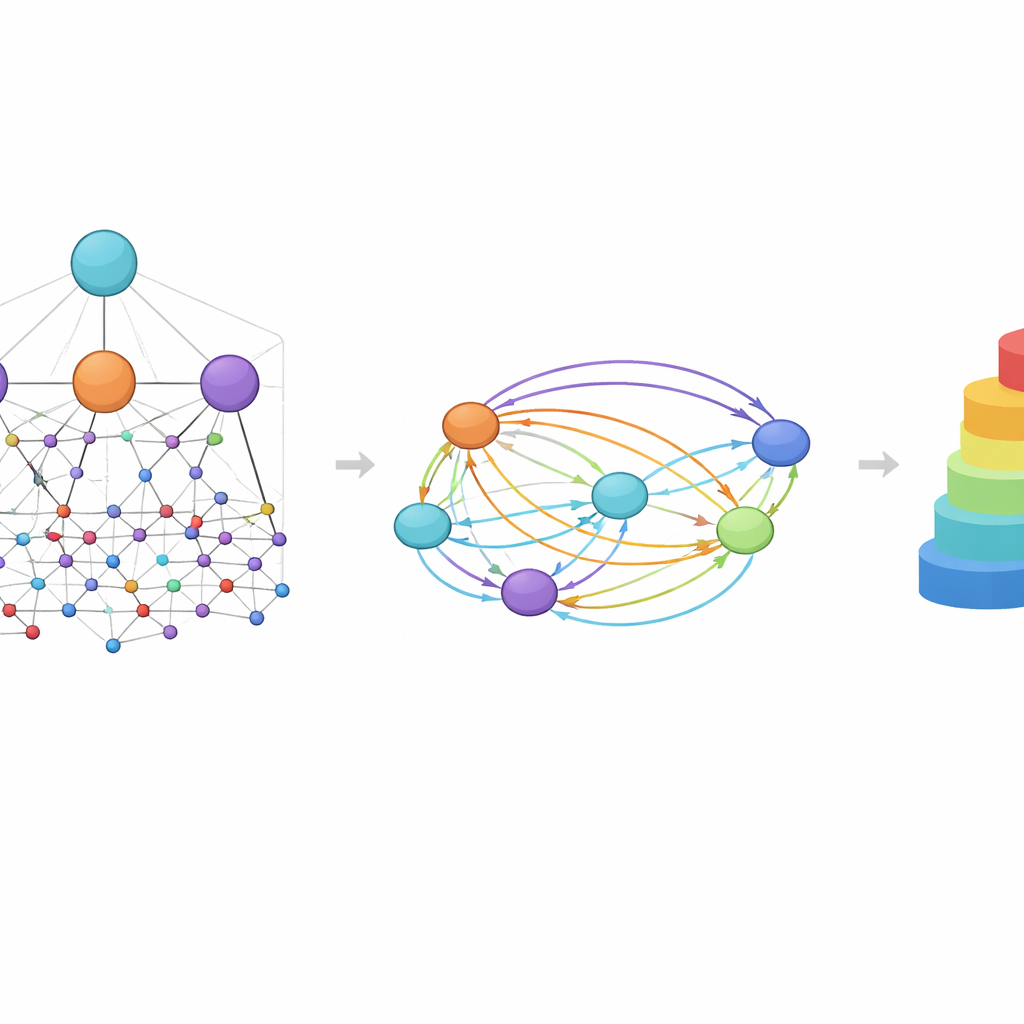
Ver la química a través de los ojos del modelo
Porque HimNet utiliza atención en múltiples niveles, sus predicciones pueden visualizarse de maneras que los químicos pueden interpretar. En estudios de caso sobre la permeabilidad de la barrera hematoencefálica, las regiones de una molécula que aumentan la penetración se resaltaron con colores cálidos, mientras que las características que la dificultan aparecieron en tonos fríos. Los anillos hidrofóbicos y los motivos fluorados tendieron a aumentar la permeabilidad, mientras que grupos muy polares, como ácidos carboxílicos o ciertos centros carbonílicos y nitrogenados, a menudo la redujeron. De forma llamativa, el modelo a veces trató dos anillos aparentemente similares de manera diferente según su entorno, reflejando cómo el contexto químico real puede convertir una parte de la molécula en una ayuda y otra en un obstáculo.
Qué significa esto para los medicamentos del futuro
Para los no especialistas, el mensaje clave es que HimNet ofrece una forma más matizada de que la IA “lea” las moléculas, no solo como listas de átomos sino como partes que interactúan cuyo comportamiento combinado determina si un compuesto tiene probabilidades de convertirse en un buen fármaco. Al capturar estas interacciones multinivel y explicar su enfoque en términos químicamente sensatos, HimNet puede ayudar a los investigadores a cribar vastas bibliotecas virtuales con mayor confianza. Aunque el método es más exigente computacionalmente y aún necesita ampliaciones para manejar la forma tridimensional completa y moléculas muy flexibles, apunta hacia herramientas más inteligentes y transparentes que podrían acortar el camino desde la idea molecular hasta un medicamento seguro y eficaz.
Cita: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Palabras clave: predicción de propiedades moleculares, redes neuronales gráficas, descubrimiento de fármacos, modelado ADMET, IA explicable