Clear Sky Science · pt
Uma rede hierárquica de mensagens de interação para previsão precisa de propriedades moleculares
Atalhos mais inteligentes para encontrar medicamentos melhores
Lançar um novo medicamento no mercado é um processo lento e caro, em parte porque químicos precisam testar um grande número de moléculas no laboratório para verificar se são seguras, eficazes e se se comportam bem no organismo. Este estudo apresenta um novo modelo de inteligência artificial, chamado HimNet, projetado para prever essas propriedades cruciais semelhantes a fármacos com muito mais precisão apenas a partir da estrutura de uma molécula, ajudando cientistas a concentrar os experimentos nos candidatos mais promissores.
Por que é tão difícil prever o comportamento das moléculas
Cada potencial fármaco deve satisfazer uma lista rigorosa conhecida como ADMET: como é absorvido, distribuído, metabolizado, excretado e se é tóxico. Modelos computacionais tradicionais dependem de regras químicas manuais ou de sistemas de aprendizado profundo que veem moléculas como redes simples de átomos e ligações. Essas abordagens antigas frequentemente deixam de captar como diferentes partes de uma molécula interagem de maneiras sutis e não aditivas — por exemplo, como dois anéis distantes podem empilhar-se, ou como um grupo polar de um lado pode atenuar o efeito de uma região apolar do outro. Como resultado, previsões sobre solubilidade, toxicidade ou penetração da barreira hematoencefálica podem ser pouco confiáveis quando químicos exploram novas regiões do espaço químico.
Olhando para as moléculas em camadas
HimNet aborda esse problema ao enxergar cada molécula em vários níveis simultaneamente. No nível mais fino, ele acompanha átomos individuais e as ligações entre eles. Um segundo nível agrupa átomos em “motivos” quimicamente significativos, como anéis aromáticos ou grupos ácidos, enquanto um terceiro nível global representa a molécula inteira. HimNet troca informações entre essas camadas usando um mecanismo hierárquico de passagem de mensagens de interação — essencialmente uma conversa estruturada na qual átomos conversam com motivos, motivos conversam entre si e tudo contribui para um quadro geral do comportamento da molécula. Em paralelo, o modelo também analisa as «impressões digitais» moleculares padrão, codificações digitais compactas amplamente utilizadas em química medicinal, e aprende quais aspectos dessas impressões são mais consistentes entre diferentes descrições da mesma molécula.
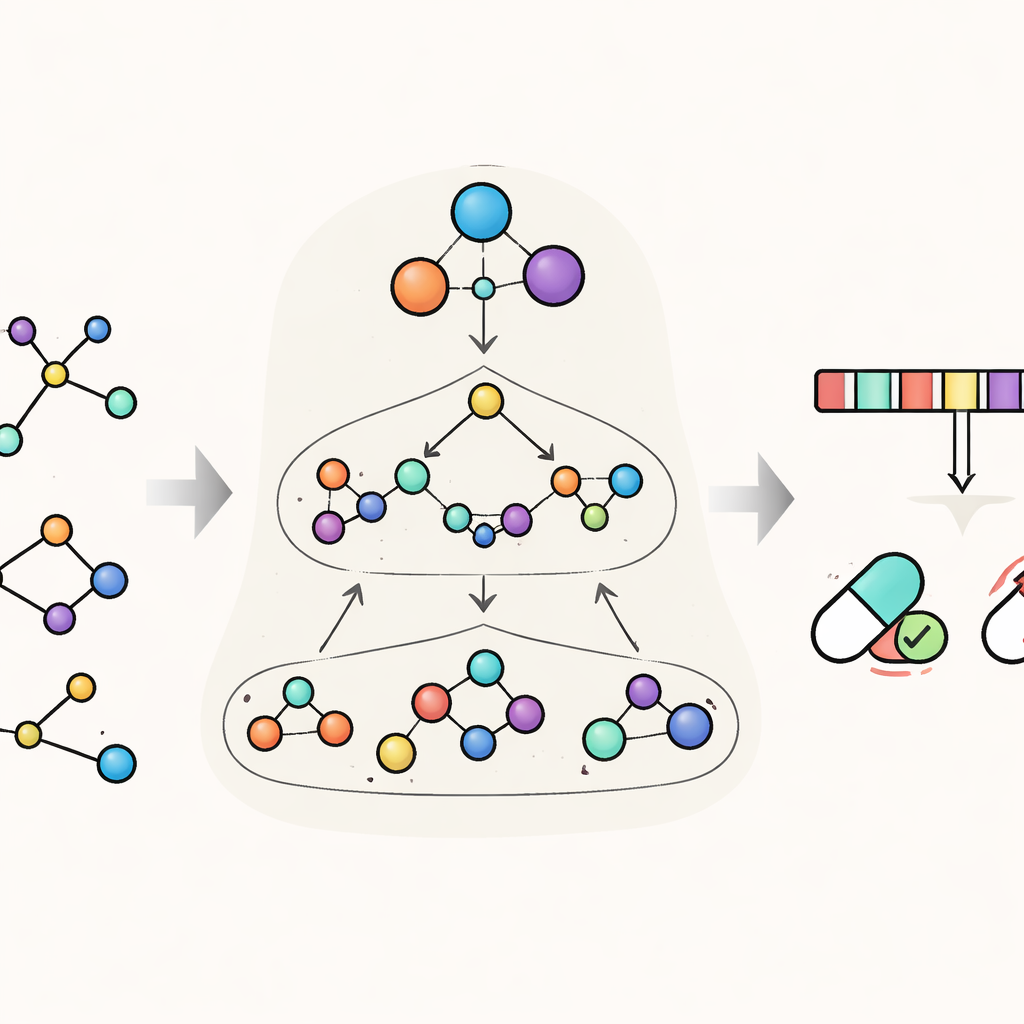
Ensinando o modelo a focar no que importa
Uma ideia central no HimNet é a atenção: o modelo aprende a destacar quais átomos e motivos são mais importantes para uma dada propriedade e como suas influências se reforçam ou se cancelam. Um caminho na rede segue cuidadosamente as ligações químicas reais, preservando os detalhes locais em que os químicos confiam. Um segundo caminho constrói “pontes” de longo alcance pela molécula, permitindo que grupos distantes interajam mesmo estando separados por várias ligações. Uma porta aprendível combina essas duas vias, equilibrando conectividade química estrita com um contexto global mais flexível. Módulos adicionais alinham a visão em camadas do grafo com a visão das impressões digitais, produzindo uma representação compacta final que captura tanto a estrutura fina quanto os padrões químicos mais amplos.
Colocando a abordagem à prova
Para avaliar a eficácia desse projeto, os autores testaram o HimNet em onze conjuntos de dados cobrindo muitas tarefas relevantes para a descoberta de fármacos. Isso inclui benchmarks clássicos para toxicidade, penetração da barreira hematoencefálica e propriedades físicas básicas como solubilidade, bem como problemas do mundo real mais exigentes, como estabilidade metabólica, atividade antimalárica e depuração hepática em diferentes espécies. Em grande parte dessas tarefas, o HimNet igualou ou superou os melhores modelos existentes, frequentemente com margem clara. Importante, o modelo permaneceu robusto mesmo em conjuntos de dados desafiadores, embora o desempenho tenha caído um pouco em coleções muito pequenas, ruidosas ou altamente desequilibradas, onde qualquer método tem dificuldades. Estudos de ablação — removendo sistematicamente partes da arquitetura — mostraram que cada componente, desde a atenção entre níveis até a fusão das impressões digitais, contribui de forma mensurável para a precisão.
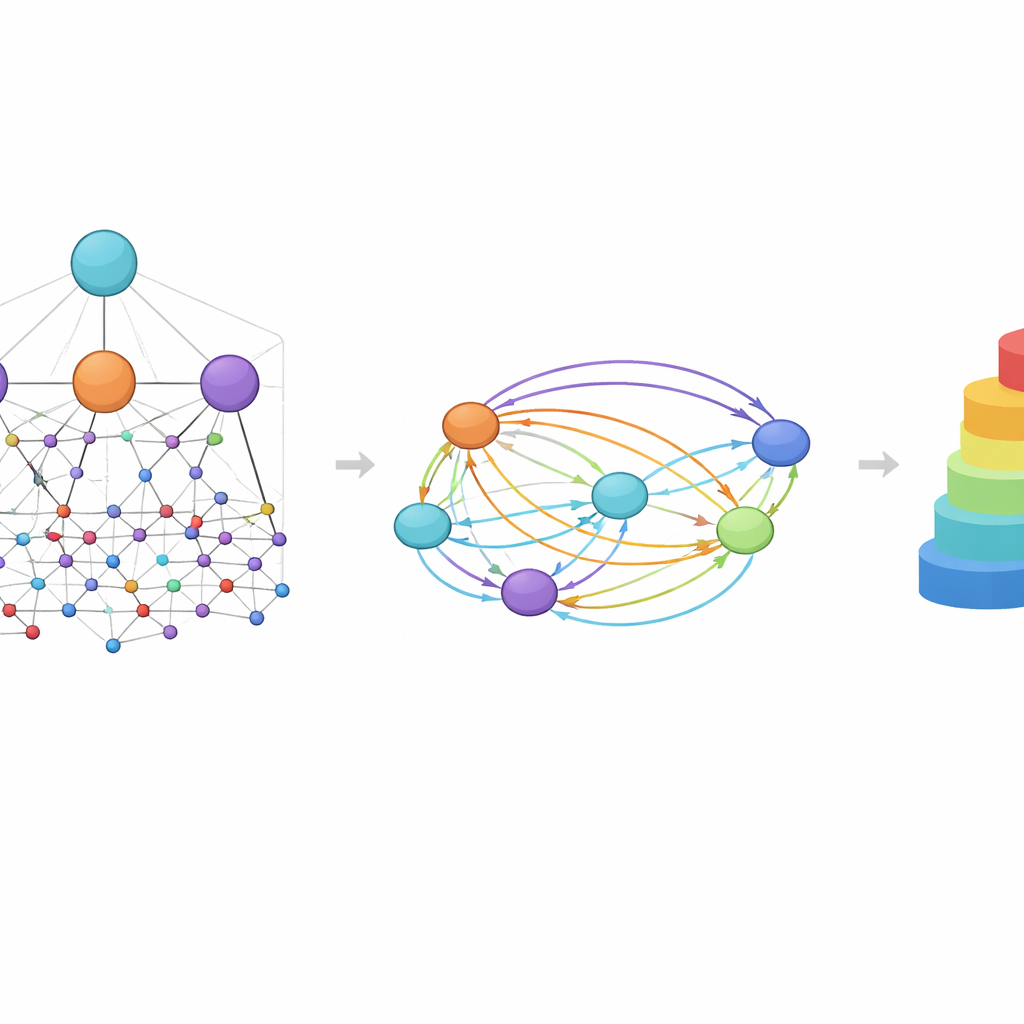
Vendo a química pelos olhos do modelo
Como o HimNet usa atenção em múltiplos níveis, suas previsões podem ser visualizadas de maneiras que químicos conseguem interpretar. Em estudos de caso sobre permeabilidade da barreira hematoencefálica, regiões da molécula que aumentam a penetração foram destacadas em cores quentes, enquanto feições que dificultam a penetração apareceram em tons frios. Anéis hidrofóbicos e motivos fluorados tenderam a aumentar a permeabilidade, enquanto grupos altamente polares, como ácidos carboxílicos ou certos centros carbonila e de nitrogênio, frequentemente a reduziram. Notavelmente, o modelo às vezes tratou dois anéis aparentemente semelhantes de forma diferente dependendo de seu entorno, refletindo como o contexto químico real pode transformar uma parte da molécula em auxílio e outra em obstáculo.
O que isso significa para medicamentos futuros
Para não especialistas, a mensagem-chave é que o HimNet oferece uma maneira mais nuançada para a IA “ler” moléculas, não apenas como listas de átomos, mas como partes que interagem cujo comportamento combinado determina se um composto tem probabilidade de ser um bom fármaco. Ao capturar essas interações multinível e explicar seu foco em termos quimicamente sensatos, o HimNet pode ajudar pesquisadores a vasculhar vastas bibliotecas virtuais com maior confiança. Embora o método seja mais exigente computacionalmente e ainda precise de extensões para lidar com forma tridimensional completa e moléculas muito flexíveis, ele aponta para ferramentas mais inteligentes e transparentes que podem encurtar o caminho da ideia molecular até um medicamento seguro e eficaz.
Citação: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Palavras-chave: previsão de propriedades moleculares, redes neurais em grafos, descoberta de medicamentos, modelagem ADMET, IA explicável