Clear Sky Science · sv
Ett hierarkiskt meddelandenätverk för exakta förutsägelser av molekylära egenskaper
Smartare genvägar för att hitta bättre läkemedel
Att föra ett nytt läkemedel till marknaden är långsamt och kostsamt, delvis eftersom kemister måste testa enorma mängder molekyler i labb för att avgöra om de är säkra, effektiva och beter sig väl i kroppen. Denna studie presenterar en ny artificiell intelligensmodell, kallad HimNet, utformad för att förutsäga dessa avgörande läkemedelsliknande egenskaper mycket mer träffsäkert utifrån en molekyls struktur ensam, vilket hjälper forskare att rikta experiment mot de mest lovande kandidaterna.
Varför det är så svårt att förutsäga molekylers beteende
Varje potentiellt läkemedel måste uppfylla en krävande checklista känd som ADMET: hur det absorberas, distribueras, metaboliseras, utsöndras och om det är giftigt. Traditionella datorbaserade modeller förlitar sig antingen på handgjorda kemiska regler eller på djupinlärningssystem som ser molekyler som enkla nätverk av atomer och bindningar. Dessa äldre tillvägagångssätt missar ofta hur olika delar av en molekyl samverkar på subtila, icke-additiva sätt — till exempel hur två avlägsna ringar kan stapla sig eller hur en polär grupp på ena sidan kan försvaga effekten av en fet fläck på andra sidan. Som en följd kan förutsägelser om löslighet, toxicitet eller penetration genom blod-hjärnbarriären bli opålitliga när kemister försöker utforska nya områden i kemiskt rum.
Att betrakta molekyler i lager
HimNet tacklar detta problem genom att betrakta varje molekyl på flera nivåer samtidigt. På den finaste nivån spårar den individuella atomer och bindningarna mellan dem. En andra nivå grupperar atomer till kemiskt meningsfulla ”motiv”, såsom aromatiska ringar eller syragrupper, medan en tredje, global nivå representerar hela molekylen. HimNet skickar information fram och tillbaka över dessa lager med en hierarkisk interaktionsmeddelandeöverföringsmekanism — i praktiken en strukturerad konversation där atomer talar med motiv, motiv talar med varandra, och allt bidrar till en helhetsbild av molekylens beteende. Parallellt analyserar modellen också standardiserade molekylära ”fingeravtryck”, kompakta digitala avkodningar som är vanligt förekommande i läkemedelskemi, och lär sig vilka aspekter av dessa fingeravtryck som är mest konsistenta över olika beskrivningar av samma molekyl.
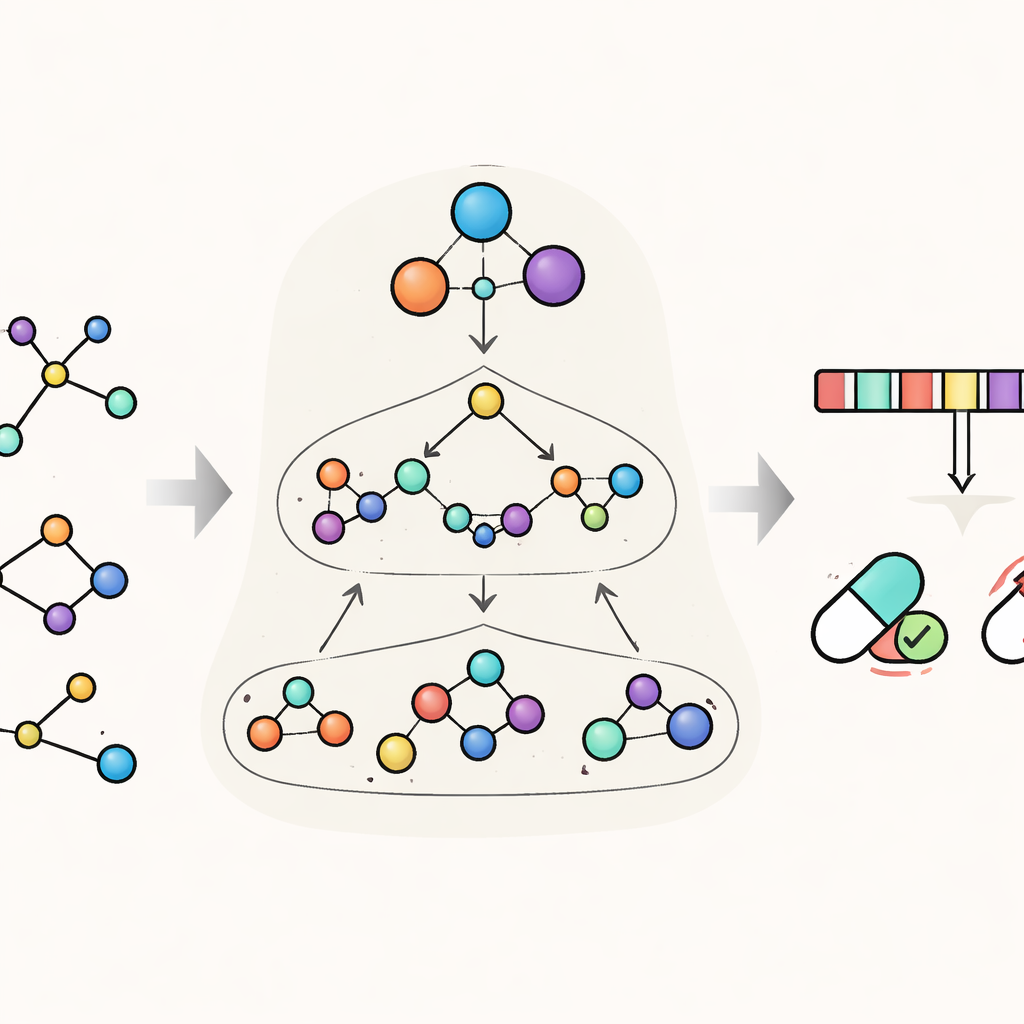
Lära modellen att fokusera på det som spelar roll
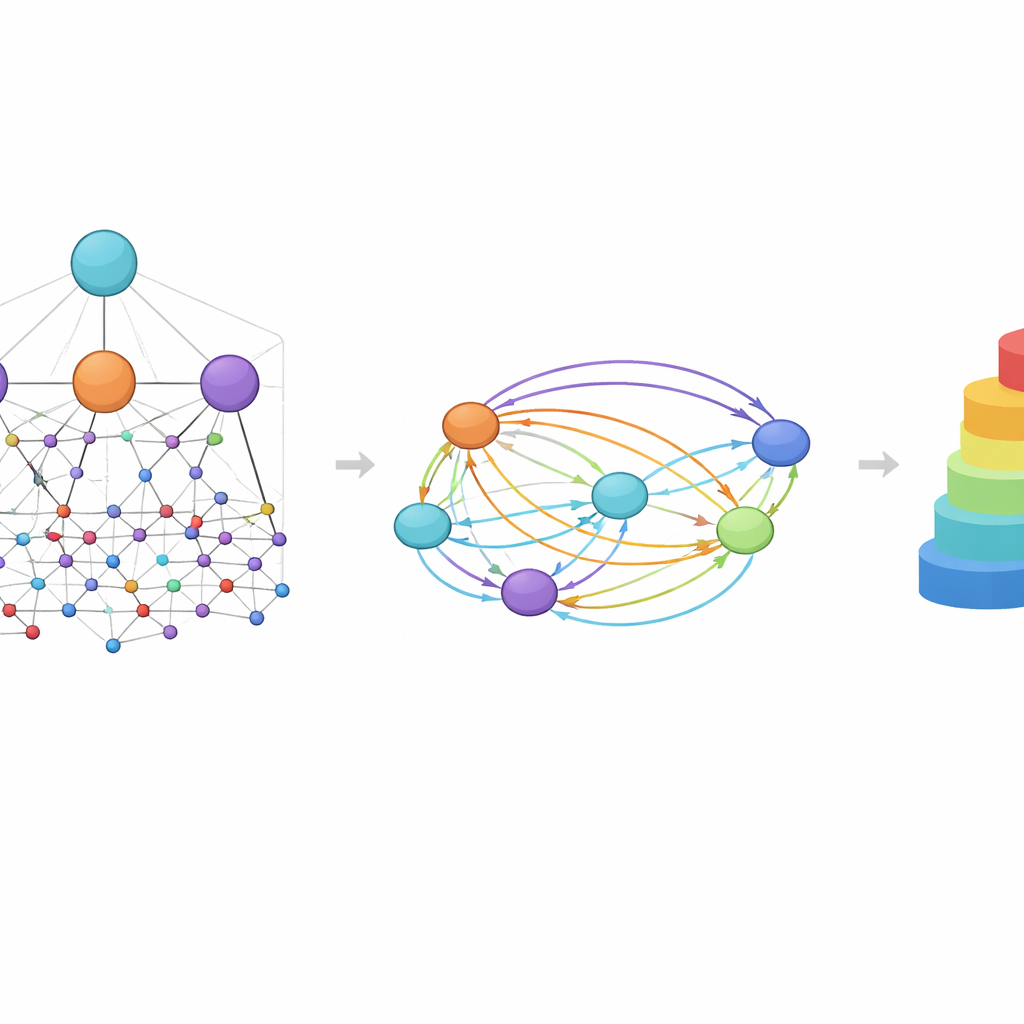
En central idé i HimNet är uppmärksamhet: modellen lär sig att lyfta fram vilka atomer och motiv som är viktigast för en viss egenskap och hur deras påverkan förstärker eller släcker ut varandra. En väg i nätverket följer noggrant de faktiska kemiska bindningarna och bevarar de lokala detaljer kemister litar på. En andra väg bygger långväga ”broar” över molekylen, vilket tillåter avlägsna grupper att interagera även om de skiljs åt av flera bindningar. En inlärbar grind blandar dessa två strömmar och balanserar strikt kemisk sammanlänkning med flexibel, global kontext. Ytterligare moduler justerar den lagerindelade grafvyn med fingeravtrycksvyn och ger en slutlig kompakt representation som fångar både den fina strukturen och de bredare kemiska mönstren.
Sätta metoden på prov
För att bedöma hur väl denna design fungerar utvärderade författarna HimNet på elva dataset som täcker många uppgifter som spelar roll i läkemedelsupptäckt. Dessa inkluderar klassiska benchmark-tester för toxicitet, penetration genom blod-hjärnbarriären och grundläggande fysikaliska egenskaper som löslighet, samt mer krävande, verkliga problem såsom metabol stabilitet, antimalariell aktivitet och leverrensning i olika arter. I de flesta av dessa uppgifter motsvarade eller överträffade HimNet de bästa befintliga modellerna, ofta med en tydlig marginal. Viktigt är att modellen förblev robust även på utmanande dataset, även om prestandan sjönk något för mycket små, brusiga eller starkt obalanserade samlingar där alla metoder har svårt. Ablationsstudier — systematiska borttaganden av delar av arkitekturen — visade att varje komponent, från tvärnivåuppmärksamhet till fingeravtrycksfusion, gör ett mätbart bidrag till noggrannheten.
Se kemi genom modellens ögon
Eftersom HimNet använder uppmärksamhet på flera nivåer kan dess förutsägelser visualiseras på sätt som kemister kan tolka. I fallstudier om penetration genom blod-hjärnbarriären markerades molekylregioner som ökar penetration i varma färger, medan funktioner som hindrar penetration visade sig i kalla färger. Hydrofoniska ringar och fluorinerade motiv tenderade att öka permeabiliteten, medan starkt polära grupper, såsom karboxylsyror eller vissa karbonyl- och kvävecentra, ofta minskade den. Slående nog behandlade modellen ibland två till synes likartade ringar olika beroende på deras omgivning, vilket speglar hur verklig kemisk kontext kan förvandla en del av en molekyl till en hjälp och en annan till ett hinder.
Vad detta betyder för framtida läkemedel
För icke-specialister är huvudbudskapet att HimNet erbjuder ett mer nyanserat sätt för AI att ”läsa” molekyler, inte bara som listor av atomer utan som interagerande delar vars samlade beteende avgör om en förening sannolikt blir ett bra läkemedel. Genom att fånga dessa flerskiktade interaktioner och förklara sitt fokus i kemiskt rimliga termer kan HimNet hjälpa forskare att sålla igenom stora virtuella bibliotek med större självförtroende. Medan metoden är mer beräkningskrävande och fortfarande behöver utvidgas för att hantera full tredimensionell form och mycket flexibla molekyler, pekar den mot smartare, mer transparenta verktyg som kan förkorta vägen från molekylär idé till säkert och effektivt läkemedel.
Citering: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Nyckelord: förutsägelse av molekylära egenskaper, grafneuronätverk, Läkemedelsupptäckt, ADMET-modellering, förklarlig AI