Clear Sky Science · fr
Un réseau de messages d’interaction hiérarchique pour une prédiction précise des propriétés moléculaires
Raccourcis plus intelligents pour trouver de meilleurs médicaments
Mettre un nouveau médicament sur le marché est lent et coûteux, en partie parce que les chimistes doivent tester en laboratoire un très grand nombre de molécules pour vérifier qu’elles sont sûres, efficaces et se comportent correctement dans l’organisme. Cette étude présente un nouveau modèle d’intelligence artificielle, appelé HimNet, conçu pour prédire ces propriétés cruciales de type médicament à partir de la seule structure d’une molécule, et ce avec bien plus de précision, aidant ainsi les scientifiques à concentrer les expériences sur les candidats les plus prometteurs.
Pourquoi il est si difficile de prédire le comportement des molécules
Chaque médicament potentiel doit satisfaire une liste d’exigences exigeante connue sous le sigle ADMET : absorption, distribution, métabolisme, excrétion et toxicité. Les modèles informatiques traditionnels reposent soit sur des règles chimiques conçues manuellement, soit sur des systèmes d’apprentissage profond qui considèrent les molécules comme de simples réseaux d’atomes et de liaisons. Ces approches plus anciennes négligent souvent la manière dont différentes parties d’une molécule interagissent de façon subtile et non additive — par exemple, comment deux cycles éloignés peuvent s’empiler, ou comment un groupe polaire d’un côté peut atténuer l’effet d’une zone lipophile de l’autre. En conséquence, les prédictions de solubilité, de toxicité ou de pénétration de la barrière hémato-encéphalique peuvent être peu fiables lorsque les chimistes explorent de nouvelles régions de l’espace chimique.
Regarder les molécules en couches
HimNet s’attaque à ce problème en considérant chaque molécule à plusieurs échelles simultanément. Au niveau le plus fin, il suit les atomes individuels et les liaisons qui les relient. Un second niveau regroupe les atomes en « motifs » chimiquement signifiants, tels que des cycles aromatiques ou des groupes acides, tandis qu’un troisième niveau global représente la molécule entière. HimNet fait transiter l’information entre ces couches au moyen d’un mécanisme hiérarchique d’échange de messages — essentiellement une conversation structurée dans laquelle les atomes communiquent avec les motifs, les motifs entre eux, et tout contribue à une image d’ensemble du comportement de la molécule. En parallèle, le modèle analyse aussi des « empreintes » moléculaires standard, encodages numériques compacts largement utilisés en chimie médicamenteuse, et apprend quelles facettes de ces empreintes sont les plus cohérentes entre différentes représentations d’une même molécule.
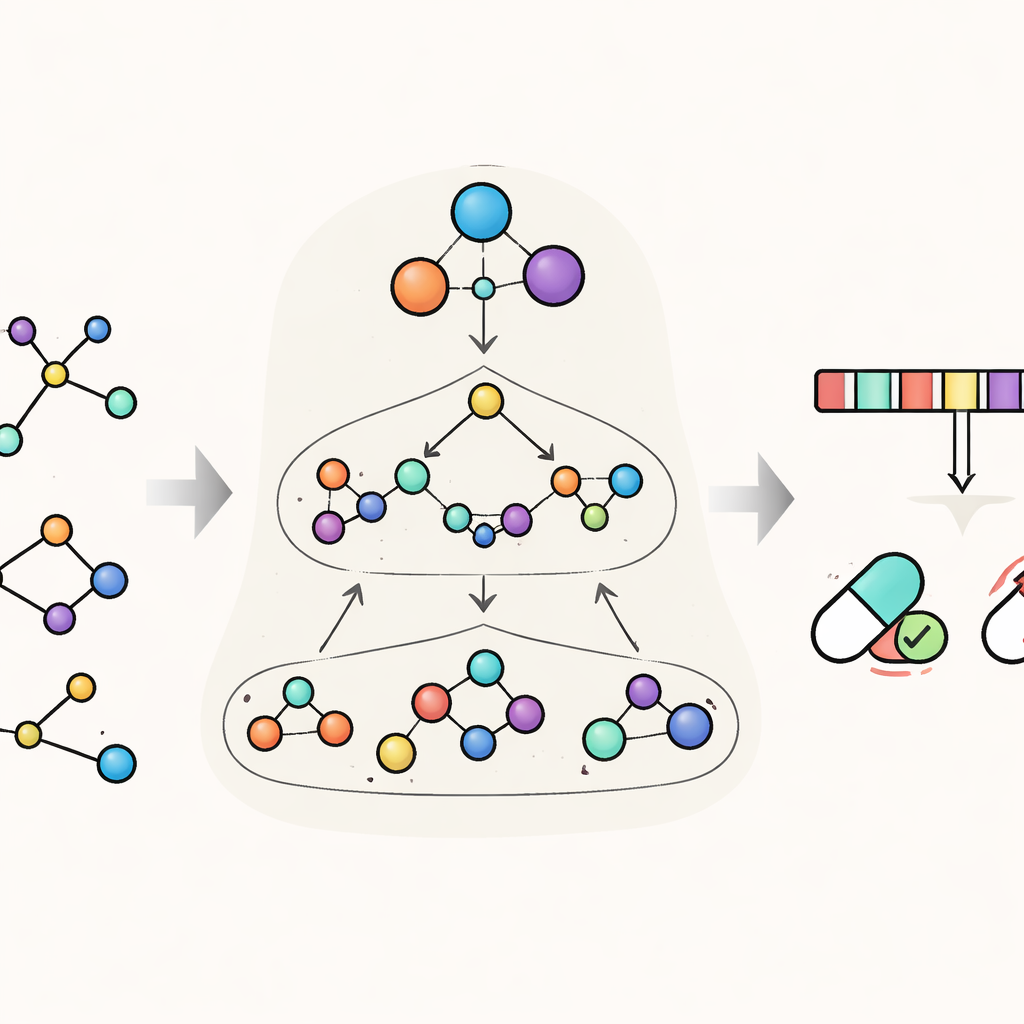
Apprendre au modèle à se concentrer sur l’essentiel
Une idée centrale de HimNet est l’attention : le modèle apprend à mettre en évidence quels atomes et quels motifs sont les plus importants pour une propriété donnée et comment leurs influences se renforcent ou s’annulent. Une branche du réseau suit attentivement les liaisons chimiques réelles, préservant les détails locaux auxquels les chimistes font confiance. Une autre branche construit des « ponts » à plus longue portée à travers la molécule, permettant à des groupes éloignés d’interagir même s’ils sont séparés par plusieurs liaisons. Une porte paramétrable combine ces deux flux, équilibrant la connectivité chimique stricte avec un contexte global plus flexible. Des modules additionnels alignent la vue en couches du graphe avec la vue par empreinte, fournissant une représentation compacte finale qui capture à la fois la structure fine et les schémas chimiques plus larges.
Mettre l’approche à l’épreuve
Pour évaluer l’efficacité de cette architecture, les auteurs ont testé HimNet sur onze jeux de données couvrant de nombreuses tâches importantes en découverte de médicaments. Cela inclut des benchmarks classiques pour la toxicité, la pénétration de la barrière hémato-encéphalique et des propriétés physiques de base comme la solubilité, ainsi que des problèmes plus exigeants et réalistes tels que la stabilité métabolique, l’activité antipaludique et l’élimination hépatique chez différentes espèces. Pour la plupart de ces tâches, HimNet a égalé ou dépassé les meilleurs modèles existants, souvent avec une marge nette. De façon importante, le modèle est resté robuste même sur des ensembles difficiles, bien que les performances aient légèrement baissé pour des collections très petites, bruyantes ou fortement déséquilibrées, où toute méthode éprouve des difficultés. Des études d’ablation — consistant à supprimer systématiquement des éléments de l’architecture — ont montré que chaque composant, de l’attention inter-niveaux à la fusion des empreintes, apporte une contribution mesurable à la précision.
Voir la chimie à travers les yeux du modèle
Parce que HimNet utilise l’attention à plusieurs niveaux, ses prédictions peuvent être visualisées de manière interprétable pour les chimistes. Dans des études de cas sur la perméabilité de la barrière hémato-encéphalique, les régions d’une molécule qui favorisent la pénétration étaient mises en valeur par des couleurs chaudes, tandis que les caractéristiques qui la gênent apparaissaient en couleurs froides. Les cycles hydrophobes et les motifs fluorés ont tendance à augmenter la perméabilité, alors que des groupes fortement polaires, tels que les acides carboxyliques ou certains centres carbonylés et azotés, la réduisent souvent. Fait frappant, le modèle a parfois traité deux cycles apparemment similaires de manière différente selon leur environnement, reflétant comment le contexte chimique réel peut transformer une partie d’une molécule en atout dans un cas et en obstacle dans un autre.
Ce que cela signifie pour les médicaments de demain
Pour les non-spécialistes, le message clé est que HimNet offre une façon plus nuancée pour l’IA de « lire » les molécules, non seulement comme des listes d’atomes, mais comme des parties en interaction dont le comportement combiné détermine si un composé a des chances de devenir un bon médicament. En capturant ces interactions multiniveaux et en expliquant ses mises en avant en termes chimiquement sensés, HimNet peut aider les chercheurs à trier d’immenses bibliothèques virtuelles avec plus de confiance. Bien que la méthode soit plus coûteuse en calcul et nécessite encore des extensions pour traiter la géométrie tridimensionnelle complète et les molécules très flexibles, elle ouvre la voie à des outils plus intelligents et plus transparents susceptibles de raccourcir le chemin de l’idée moléculaire au médicament sûr et efficace.
Citation: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Mots-clés: prédiction des propriétés moléculaires, réseaux de neurones à graphes, découverte de médicaments, modélisation ADMET, IA explicable