Clear Sky Science · nl
Een hiërarchisch interactie-berichtennet voor nauwkeurige voorspelling van moleculaire eigenschappen
Slimmere snelkoppelingen om betere medicijnen te vinden
Het op de markt brengen van een nieuw geneesmiddel is traag en duur, deels omdat chemici in het laboratorium enorme aantallen moleculen moeten testen om te bepalen of ze veilig, effectief en goed gedragend in het lichaam zijn. Deze studie introduceert een nieuw kunstmatig-intelligentie model, HimNet genoemd, dat is ontworpen om deze cruciale medicijnachtige eigenschappen veel nauwkeuriger te voorspellen op basis van alleen de structuur van een molecuul, zodat onderzoekers experimenten kunnen richten op de meest veelbelovende kandidaten.
Waarom het voorspellen van molecuulgedrag zo moeilijk is
Elk potentieel geneesmiddel moet voldoen aan een veeleisende checklist die bekendstaat als ADMET: hoe het wordt geabsorbeerd, verdeeld, gemetaboliseerd, uitgescheiden en of het toxisch is. Traditionele computermodellen vertrouwen ofwel op handgemaakte chemische regels of op deep-learning systemen die moleculen zien als eenvoudige netwerken van atomen en bindingen. Deze oudere benaderingen missen vaak hoe verschillende delen van een molecuul op subtiele, niet-additieve manieren samenwerken—bijvoorbeeld hoe twee verre ringen kunnen stapelen, of hoe een polaire groep aan de ene kant het effect van een vettig oppervlak aan de andere kant kan verzwakken. Daardoor kunnen voorspellingen over oplosbaarheid, toxiciteit of doorgang van de bloed-hersenbarrière onbetrouwbaar zijn wanneer chemici nieuwe gebieden van de chemische ruimte verkennen.
Moleculen in lagen bekijken
HimNet pakt dit probleem aan door elk molecuul op meerdere niveaus tegelijk te bekijken. Op het fijnste niveau volgt het individuele atomen en de bindingen daartussen. Een tweede niveau groepeert atomen in chemisch betekenisvolle “motieven”, zoals aromatische ringen of zure groepen, terwijl een derde, globaal niveau het hele molecuul representeert. HimNet wisselt informatie uit tussen deze lagen met een hiërarchisch interactie-berichtdoorgeefmechanisme—in wezen een gestructureerd gesprek waarin atomen met motieven praten, motieven onderling communiceren en alles bijdraagt aan een totaalbeeld van het molecuulgedrag. Parallel analyseert het model ook standaard moleculaire “vingerafdrukken”, compacte digitale coderingen die veel worden gebruikt in de medicinale chemie, en leert welke aspecten van deze vingerafdrukken het meest consistent zijn tussen verschillende beschrijvingen van hetzelfde molecuul.
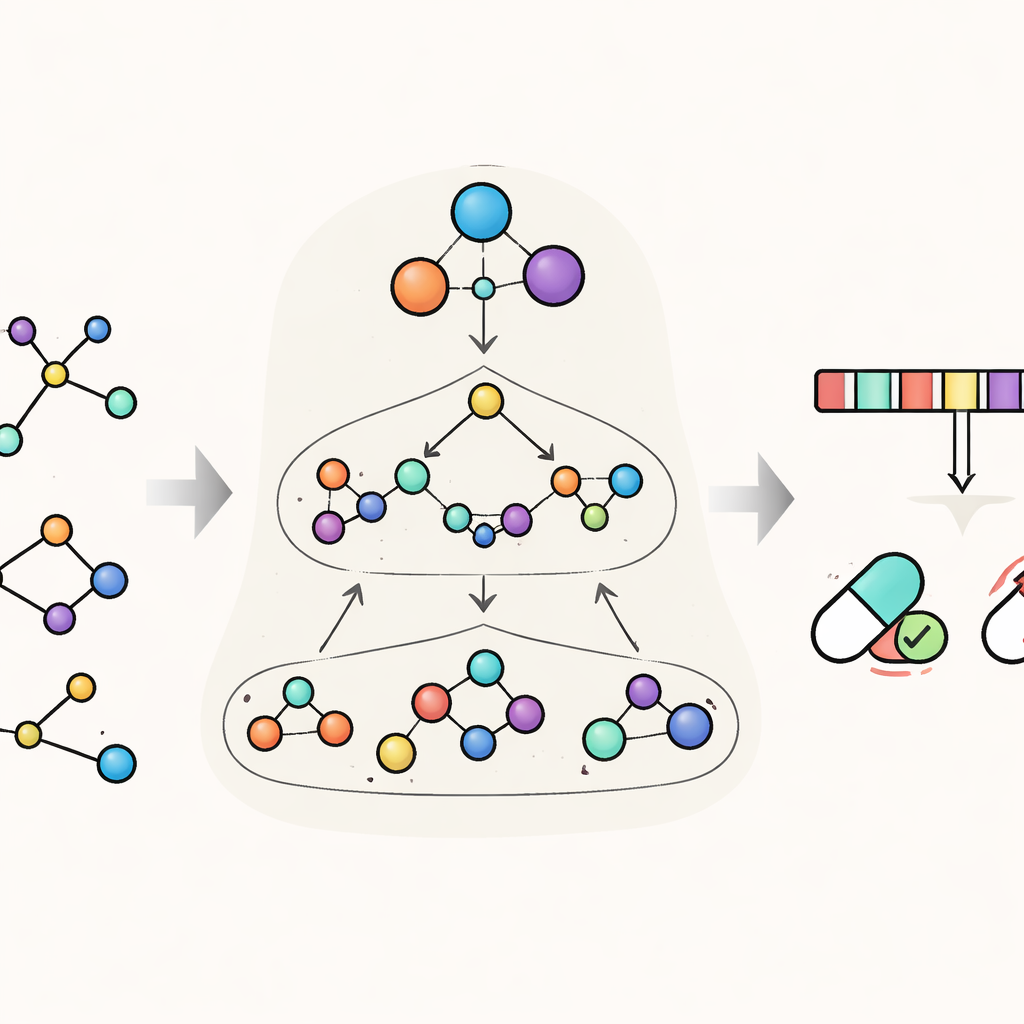
Het model leren te focussen op wat belangrijk is
Een centraal idee in HimNet is aandacht: het model leert te benadrukken welke atomen en motieven het belangrijkst zijn voor een bepaalde eigenschap en hoe hun invloeden elkaar versterken of elkaar tenietdoen. Een pad in het netwerk volgt zorgvuldig de werkelijke chemische bindingen en behoudt de lokale details waarop chemici vertrouwen. Een tweede pad bouwt langafstandige “bruggen” over het molecuul, waardoor verre groepen kunnen interageren zelfs als ze door meerdere bindingen gescheiden zijn. Een leerbare poort mengt deze twee stromen, waarbij strikte chemische connectiviteit in balans wordt gebracht met flexibele, globale context. Extra modules stemmen de gelaagde grafiekweergave af op de vingerafdrukweergave, wat een eindreductie oplevert die zowel de fijne structuur als de bredere chemische patronen vastlegt.
De aanpak op de proef stellen
Om te zien hoe goed dit ontwerp werkt, evalueerden de auteurs HimNet op elf datasets die vele taken bestrijken die belangrijk zijn in de geneesmiddelontdekking. Deze omvatten klassieke benchmarks voor toxiciteit, doorgang van de bloed-hersenbarrière en basale fysische eigenschappen zoals oplosbaarheid, evenals meer veeleisende, real-world problemen zoals metabole stabiliteit, antimalaria-activiteit en leverclearance in verschillende soorten. Voor de meeste van deze taken behaalde HimNet resultaten die gelijk waren aan of beter waren dan de beste bestaande modellen, vaak met een duidelijke marge. Belangrijk is dat het model robuust bleef, ook op uitdagende datasets, hoewel de prestaties iets daalden voor zeer kleine, lawaaierige of sterk onevenwichtige verzamelingen waar elke methode moeite mee heeft. Ablatiestudies—systematisch delen van de architectuur verwijderen—toonden aan dat elk component, van cross-level aandacht tot vingerafdrukfusie, een meetbare bijdrage levert aan de nauwkeurigheid.
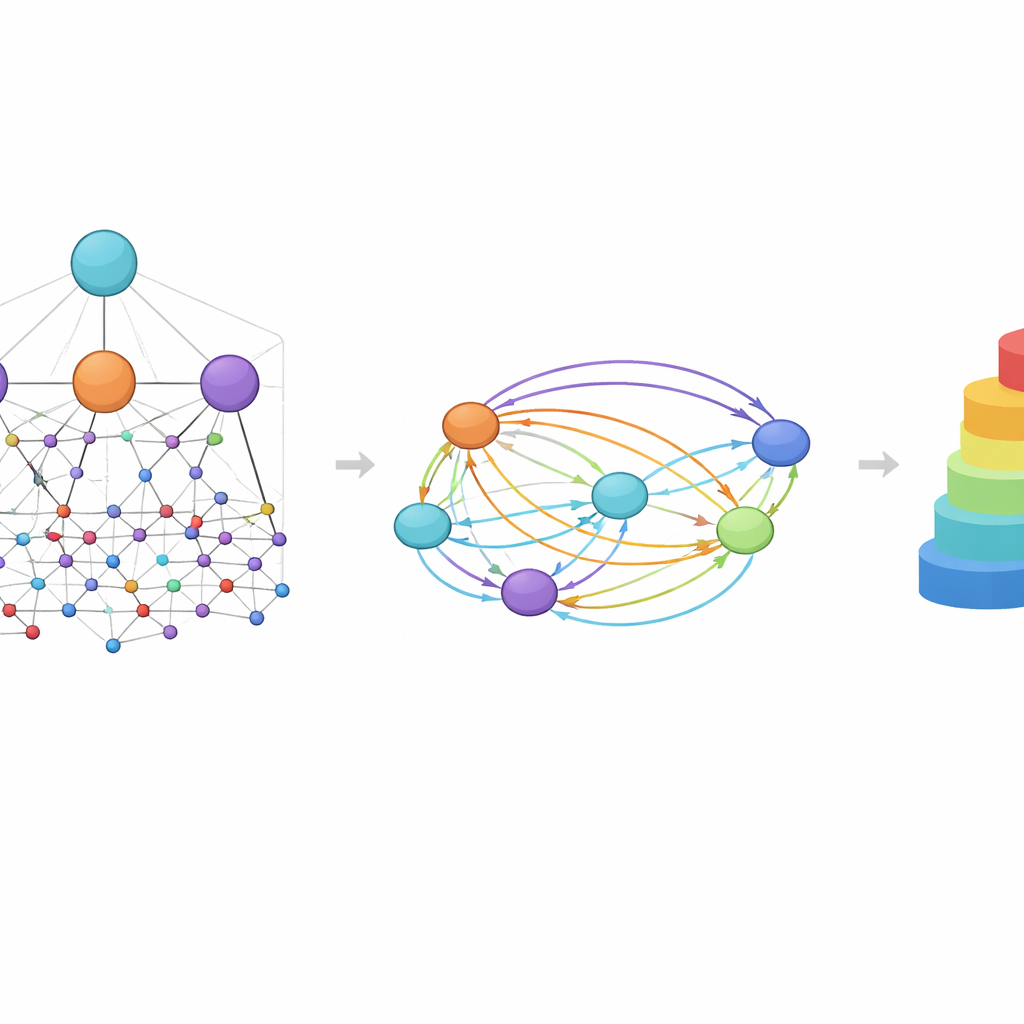
De chemie zien door de ogen van het model
Omdat HimNet aandacht op meerdere niveaus gebruikt, kunnen zijn voorspellingen worden gevisualiseerd op manieren die chemici kunnen interpreteren. In case-studies over doorgang van de bloed-hersenbarrière werden regio’s van een molecuul die penetratie vergroten in warme kleuren gemarkeerd, terwijl kenmerken die penetratie belemmeren koel verschenen. Hydrofobe ringen en gefluoreerde motieven vergrootten vaak de permeabiliteit, terwijl sterk polaire groepen, zoals carbonzuren of bepaalde carbonyl- en stikstofcentra, die vaak verminderden. Opvallend was dat het model soms twee schijnbaar vergelijkbare ringen verschillend behandelde, afhankelijk van hun omgeving, wat weerspiegelt hoe echte chemische context het ene deel van een molecuul tot hulp en het andere tot een belemmering kan maken.
Wat dit betekent voor toekomstige medicijnen
Voor niet-specialisten is de kernboodschap dat HimNet een genuanceerdere manier biedt voor AI om moleculen te “lezen”: niet alleen als lijsten met atomen, maar als interactieve onderdelen waarvan het gecombineerde gedrag bepaalt of een verbinding waarschijnlijk een goed geneesmiddel wordt. Door deze meerlaagse interacties vast te leggen en zijn focus in chemisch zinnige termen te verklaren, kan HimNet onderzoekers helpen met meer vertrouwen door enorme virtuele bibliotheken te zoeken. Hoewel de methode berekeningsintensiever is en nog uitbreidingen nodig heeft om volledige driedimensionale vormen en zeer flexibele moleculen te behandelen, wijst het de weg naar slimmerere, meer transparante hulpmiddelen die het traject van moleculair idee naar veilig en effectief geneesmiddel kunnen verkorten.
Bronvermelding: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Trefwoorden: voorspelling van moleculaire eigenschappen, graph neural networks, medicijnontdekking, ADMET-modellering, verklaarbare AI