Clear Sky Science · it
Una rete gerarchica a messaggi di interazione per una previsione accurata delle proprietà molecolari
Scorciatoie più intelligenti per trovare medicine migliori
Portare un nuovo farmaco sul mercato è lento e costoso, in parte perché i chimici devono testare in laboratorio un numero enorme di molecole per valutare sicurezza, efficacia e comportamento nell’organismo. Questo studio presenta un nuovo modello di intelligenza artificiale, chiamato HimNet, progettato per prevedere queste proprietà cruciali simili ai farmaci con molta più precisione a partire solo dalla struttura della molecola, aiutando gli scienziati a concentrare gli esperimenti sui candidati più promettenti.
Perché prevedere il comportamento delle molecole è così difficile
Ogni potenziale farmaco deve soddisfare un elenco esigente noto come ADMET: assorbimento, distribuzione, metabolismo, escrezione e tossicità. I modelli informatici tradizionali si basano o su regole chimiche costruite a mano o su sistemi di deep learning che rappresentano le molecole come semplici reti di atomi e legami. Questi approcci più vecchi spesso non colgono come diverse parti di una molecola interagiscano in modi sottili e non additivi — per esempio, come due anelli distanti possano impilarsi, o come un gruppo polare da una parte possa attenuare l’effetto di una zona idrofobica dall’altra. Di conseguenza, le previsioni su solubilità, tossicità o penetrazione della barriera emato-encefalica possono risultare inaffidabili quando i chimici esplorano nuove regioni dello spazio chimico.
Osservare le molecole a strati
HimNet affronta questo problema considerando ogni molecola su più livelli contemporaneamente. Al livello più fine, traccia i singoli atomi e i legami tra di essi. Un secondo livello raggruppa gli atomi in “motivi” chimicamente significativi, come anelli aromatici o gruppi acidi, mentre un terzo livello globale rappresenta l’intera molecola. HimNet scambia informazioni tra questi strati usando un meccanismo gerarchico di propagazione di messaggi di interazione — essenzialmente una conversazione strutturata in cui gli atomi parlano ai motivi, i motivi comunicano tra loro e tutto contribuisce a un quadro complessivo del comportamento della molecola. Parallelamente, il modello analizza anche le cosiddette “impronte” molecolari standard, codifiche digitali compatte ampiamente usate in chimica farmaceutica, e impara quali aspetti di queste impronte risultano più coerenti attraverso descrizioni diverse della stessa molecola.
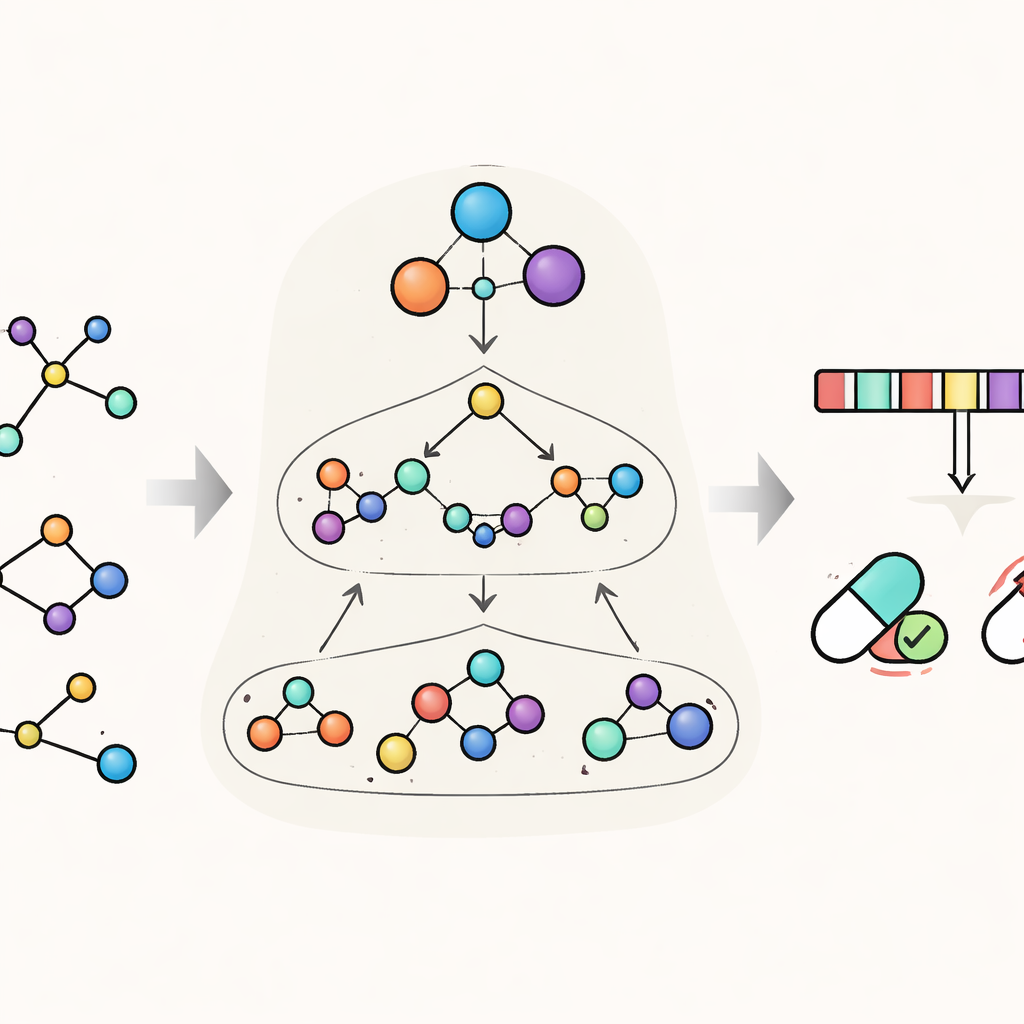
Insegnare al modello a concentrarsi su ciò che conta
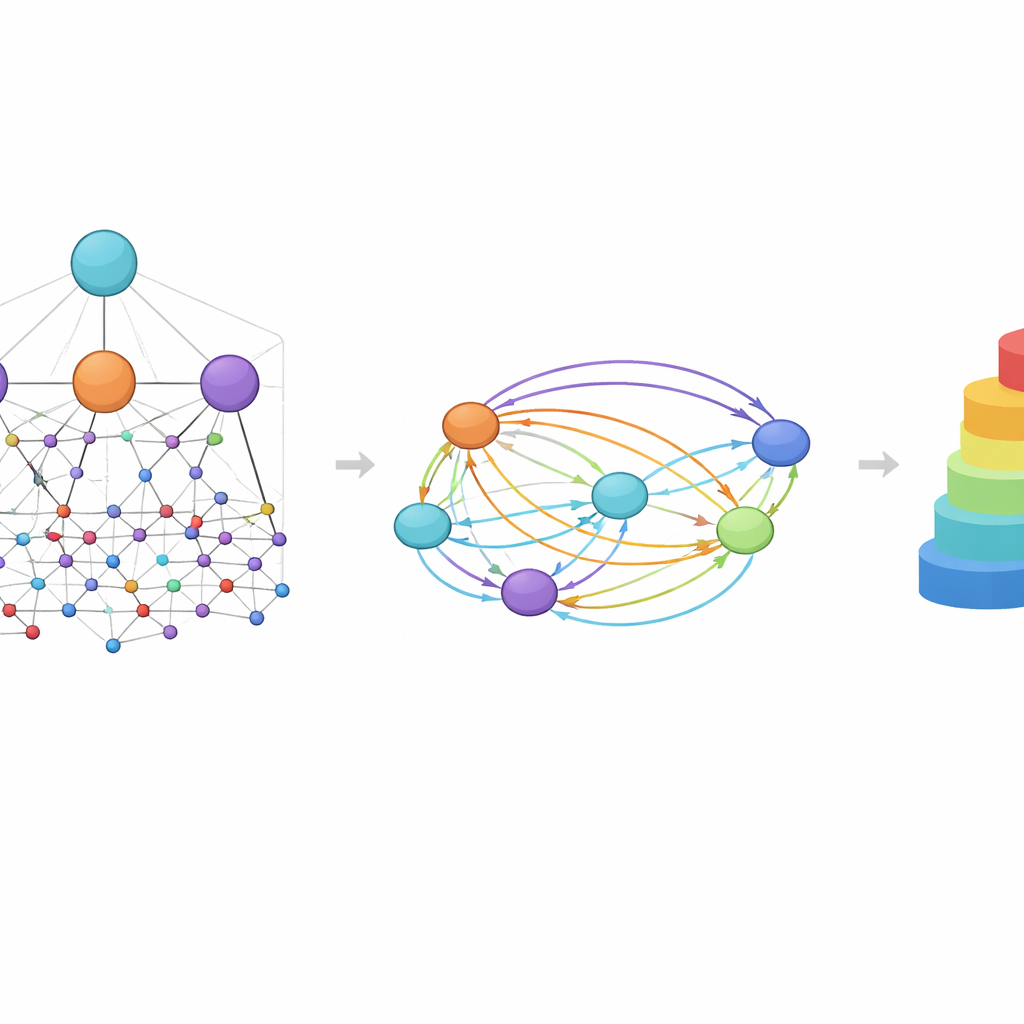
Un’idea centrale in HimNet è l’attenzione: il modello impara a evidenziare quali atomi e motivi sono più importanti per una data proprietà e come le loro influenze si rinforzano o si annullano a vicenda. Un percorso nella rete segue attentamente i legami chimici effettivi, preservando i dettagli locali di cui i chimici si fidano. Un secondo percorso costruisce “ponti” a lungo raggio attraverso la molecola, permettendo a gruppi distanti di interagire anche se separati da diversi legami. Una porta apprendente miscela questi due flussi, bilanciando la connettività chimica rigorosa con un contesto globale più flessibile. Moduli aggiuntivi allineano la vista a livelli con la vista delle impronte, fornendo una rappresentazione finale compatta che cattura sia la struttura fine sia i pattern chimici più ampi.
Mettere l’approccio alla prova
Per valutare l’efficacia di questo progetto, gli autori hanno testato HimNet su undici dataset che coprono molteplici compiti rilevanti per la scoperta di farmaci. Questi includono benchmark classici per tossicità, penetrazione della barriera emato-encefalica e proprietà fisiche di base come la solubilità, oltre a problemi più impegnativi e di interesse pratico come la stabilità metabolica, l’attività antimalarica e la clearance epatica in diverse specie. In buona parte di questi compiti, HimNet ha pareggiato o superato i migliori modelli esistenti, spesso con margini chiari. È importante notare che il modello è rimasto robusto anche su set di dati difficili, sebbene le prestazioni siano calate in misura ridotta per raccolte molto piccole, rumorose o fortemente sbilanciate, in cui qualsiasi metodo fatica. Studi di ablazione — rimuovendo sistematicamente parti dell’architettura — hanno mostrato che ogni componente, dall’attenzione cross-livello alla fusione con le impronte, contribuisce in modo misurabile all’accuratezza.
Vedere la chimica attraverso gli occhi del modello
Poiché HimNet utilizza attenzione a più livelli, le sue predizioni possono essere visualizzate in modo interpretabile per i chimici. In studi di caso sulla permeabilità della barriera emato-encefalica, le regioni di una molecola che aumentano la penetrazione sono state evidenziate con colori caldi, mentre le caratteristiche che la ostacolano apparivano in tonalità fredde. Anelli idrofobici e motivi fluorurati tendevano ad aumentare la permeabilità, mentre gruppi altamente polari, come gli acidi carbossilici o certi centri carbonilici e azotati, spesso la riducevano. In modo sorprendente, il modello talvolta ha trattato due anelli apparentemente simili in modo diverso a seconda del contesto circostante, rispecchiando come il reale contesto chimico possa trasformare una parte della molecola in un aiuto o in un ostacolo.
Cosa significa questo per i futuri farmaci
Per i non specialisti, il messaggio chiave è che HimNet offre un modo più sfumato per l’IA di “leggere” le molecole, non solo come elenchi di atomi ma come parti che interagiscono il cui comportamento combinato determina se un composto ha buone probabilità di diventare un farmaco. Catturando queste interazioni multilivello e spiegando la propria attenzione in termini chimicamente sensati, HimNet può aiutare i ricercatori a setacciare vaste librerie virtuali con maggiore fiducia. Sebbene il metodo richieda più risorse computazionali e necessiti ancora di estensioni per gestire la forma tridimensionale completa e molecole molto flessibili, indica la strada verso strumenti più intelligenti e trasparenti che potrebbero accorciare il percorso dall’idea molecolare a un medicinale sicuro ed efficace.
Citazione: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Parole chiave: predizione delle proprietà molecolari, reti neurali a grafi, scoperta di farmaci, modellizzazione ADMET, IA interpretabile