Clear Sky Science · pl
Hierarchiczna sieć komunikatów interakcyjnych do dokładnego przewidywania właściwości molekularnych
Mądrzejsze skróty do znajdowania lepszych leków
Wprowadzenie nowego leku na rynek jest powolne i kosztowne, częściowo dlatego, że chemicy muszą badać w laboratorium ogromne liczby cząsteczek, by sprawdzić, czy są bezpieczne, skuteczne i właściwie zachowują się w organizmie. W tym badaniu wprowadzono nowy model sztucznej inteligencji, zwany HimNet, zaprojektowany do znacznie dokładniejszego przewidywania tych kluczowych właściwości podobnych do leków wyłącznie na podstawie struktury cząsteczki, co pomaga naukowcom skupić eksperymenty na najbardziej obiecujących kandydatach.
Dlaczego przewidywanie zachowania cząsteczek jest takie trudne
Każdy potencjalny lek musi spełniać wymagającą listę kontrolną znaną jako ADMET: jak jest wchłaniany, dystrybuowany, metabolizowany, wydalany i czy jest toksyczny. Tradycyjne modele komputerowe albo opierają się na ręcznie opracowanych zasadach chemicznych, albo na systemach głębokiego uczenia, które traktują molekuły jako proste sieci atomów i wiązań. Te starsze podejścia często nie uwzględniają, jak różne części cząsteczki współdziałają w subtelny, nieaddytywny sposób — na przykład jak dwa odległe pierścienie mogą się układać względem siebie albo jak grupa polarna po jednej stronie może osłabić efekt tłustej plamy po drugiej. W rezultacie przewidywania dotyczące rozpuszczalności, toksyczności czy przenikania bariery krew–mózg mogą być zawodnie, gdy chemicy próbują eksplorować nowe rejony przestrzeni chemicznej.
Patrząc na cząsteczki warstwami
HimNet rozwiązuje ten problem, oglądając każdą cząsteczkę na kilku poziomach jednocześnie. Na najdrobniejszym poziomie śledzi pojedyncze atomy i wiążące je wiązania. Drugi poziom grupuje atomy w chemicznie sensowne „motywy”, takie jak pierścienie aromatyczne czy grupy kwasowe, podczas gdy trzeci, globalny poziom reprezentuje całą cząsteczkę. HimNet przekazuje informacje tam i z powrotem między tymi warstwami za pomocą hierarchicznego mechanizmu przekazywania komunikatów — w zasadzie uporządkowanej rozmowy, w której atomy rozmawiają z motywami, motywy komunikują się między sobą, a wszystko przyczynia się do ogólnego obrazu zachowania cząsteczki. Równolegle model analizuje także standardowe „odciski palców” molekularne, zwarte cyfrowe kodowania powszechnie używane w chemii leczniczej, i uczy się, które aspekty tych odcisków są najbardziej spójne w różnych opisach tej samej cząsteczki.
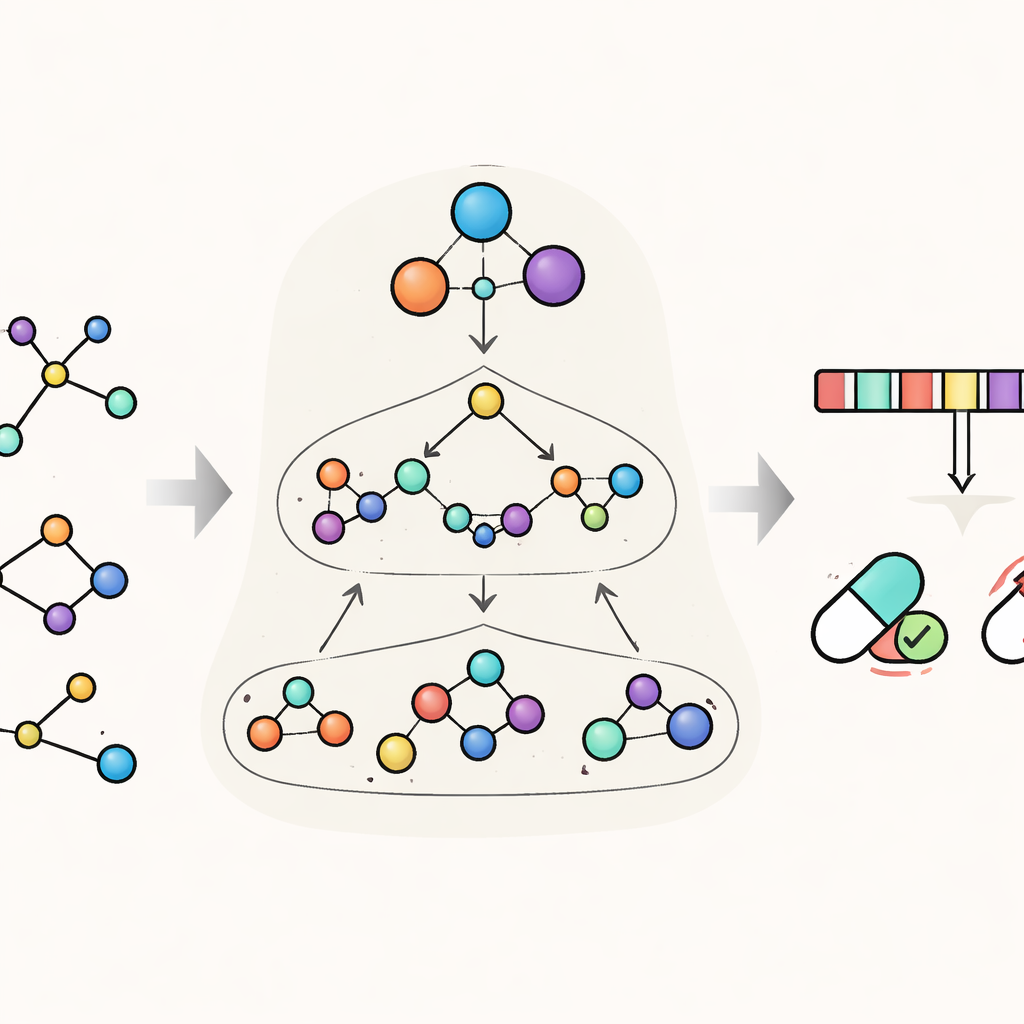
Nauczanie modelu, na czym skupić uwagę
Centralną ideą HimNet jest mechanizm uwagi: model uczy się podkreślać, które atomy i motywy są najważniejsze dla danej właściwości oraz jak ich wpływy się wzmacniają lub znoszą nawzajem. Jedna ścieżka w sieci uważnie podąża za rzeczywistymi wiązaniami chemicznymi, zachowując lokalne szczegóły, którym ufają chemicy. Druga ścieżka buduje długozasięgowe „mosty” przez cząsteczkę, pozwalając odległym grupom oddziaływać, nawet jeśli dzieli je kilka wiązań. Uczona bramka łączy te dwa strumienie, bilansując ścisłą łączność chemiczną z elastycznym, globalnym kontekstem. Dodatkowe moduły wyrównują widok grafu warstwowego z widokiem odcisków palców, dając ostateczną zwartą reprezentację, która uchwyca zarówno drobną strukturę, jak i szersze wzorce chemiczne.
Sprawdzanie podejścia w praktyce
Aby ocenić skuteczność tego projektu, autorzy przetestowali HimNet na jedenastu zestawach danych obejmujących wiele zadań istotnych w odkrywaniu leków. Obejmowały one klasyczne benchmarki dotyczące toksyczności, przenikania bariery krew–mózg i podstawowych właściwości fizycznych, takich jak rozpuszczalność, a także bardziej wymagające, rzeczywiste problemy, takie jak stabilność metaboliczna, aktywność przeciwmalaryczna i klirens wątroby u różnych gatunków. W większości tych zadań HimNet dorównywał lub przewyższał najlepsze istniejące modele, często z wyraźną przewagą. Co ważne, model pozostał odporny nawet na trudnych zestawach danych, choć wydajność nieco spadała dla bardzo małych, zaszumionych lub silnie niezrównoważonych zbiorów, z którymi każde podejście ma problemy. Badania ablacyjne — systematyczne usuwanie części architektury — wykazały, że każdy komponent, od uwagi między poziomami po fuzję odcisków palców, wnosi mierzalny wkład w dokładność.
Widzenie chemii oczami modelu
Dzięki stosowaniu uwagi na wielu poziomach przewidywania HimNet można wizualizować w sposób zrozumiały dla chemików. W studiach przypadków dotyczących przepuszczalności bariery krew–mózg obszary cząsteczki zwiększające przenikanie były podświetlane ciepłymi kolorami, podczas gdy cechy utrudniające przenikanie ukazywano w kolorach chłodnych. Pierścienie hydrofobowe i motywy fluorowane miały tendencję do zwiększania przepuszczalności, podczas gdy silnie polarne grupy, takie jak kwasy karboksylowe czy niektóre centra karbonylowe i azotowe, często ją zmniejszały. Co zaskakujące, model czasami traktował dwa pozornie podobne pierścienie inaczej w zależności od ich otoczenia, odzwierciedlając to, jak rzeczywisty kontekst chemiczny może przemienić jedną część cząsteczki w korzyść, a inną w przeszkodę.
Co to oznacza dla przyszłych leków
Dla osób spoza specjalizacji kluczowy przekaz jest taki, że HimNet oferuje bardziej subtelny sposób, w jaki AI „czyta” cząsteczki — nie tylko jako listę atomów, lecz jako współdziałające części, których łączne zachowanie decyduje o tym, czy związek ma szansę stać się dobrym lekiem. Poprzez uchwycenie tych wielowarstwowych interakcji i wyjaśnianie swojego skupienia w sensownych chemicznie terminach, HimNet może pomóc badaczom przesiewać ogromne wirtualne biblioteki z większą pewnością. Choć metoda jest bardziej wymagająca obliczeniowo i wymaga dalszych rozszerzeń, by uwzględnić pełny trójwymiarowy kształt i bardzo elastyczne cząsteczki, wskazuje kierunek ku mądrzejszym, bardziej przejrzystym narzędziom, które mogą skrócić drogę od pomysłu molekularnego do bezpiecznego i skutecznego leku.
Cytowanie: Hong, H., Wu, X., Sun, H. et al. A hierarchical interaction message net for accurate molecular property prediction. Commun Chem 9, 150 (2026). https://doi.org/10.1038/s42004-026-01922-x
Słowa kluczowe: przewidywanie właściwości molekularnych, grafowe sieci neuronowe, odkrywanie leków, modelowanie ADMET, wyjaśnialna sztuczna inteligencja