Clear Sky Science · zh
从图论到化学信息学:改良的基于键的指标与基于假设的多任务QSAR/QSPR基准
为何微小的分子连结重要
化学家常把分子比作微型城市:原子是建筑,化学键是道路。几十年来,用于预测分子行为的大多数数学工具更侧重于计算“建筑”处发生的事情,而非它们之间的“道路”。本文提出了一个简单但有力的问题:如果我们更加关注键本身,这一额外细节能否帮助计算机更好地预测潜在抗菌药物的行为?
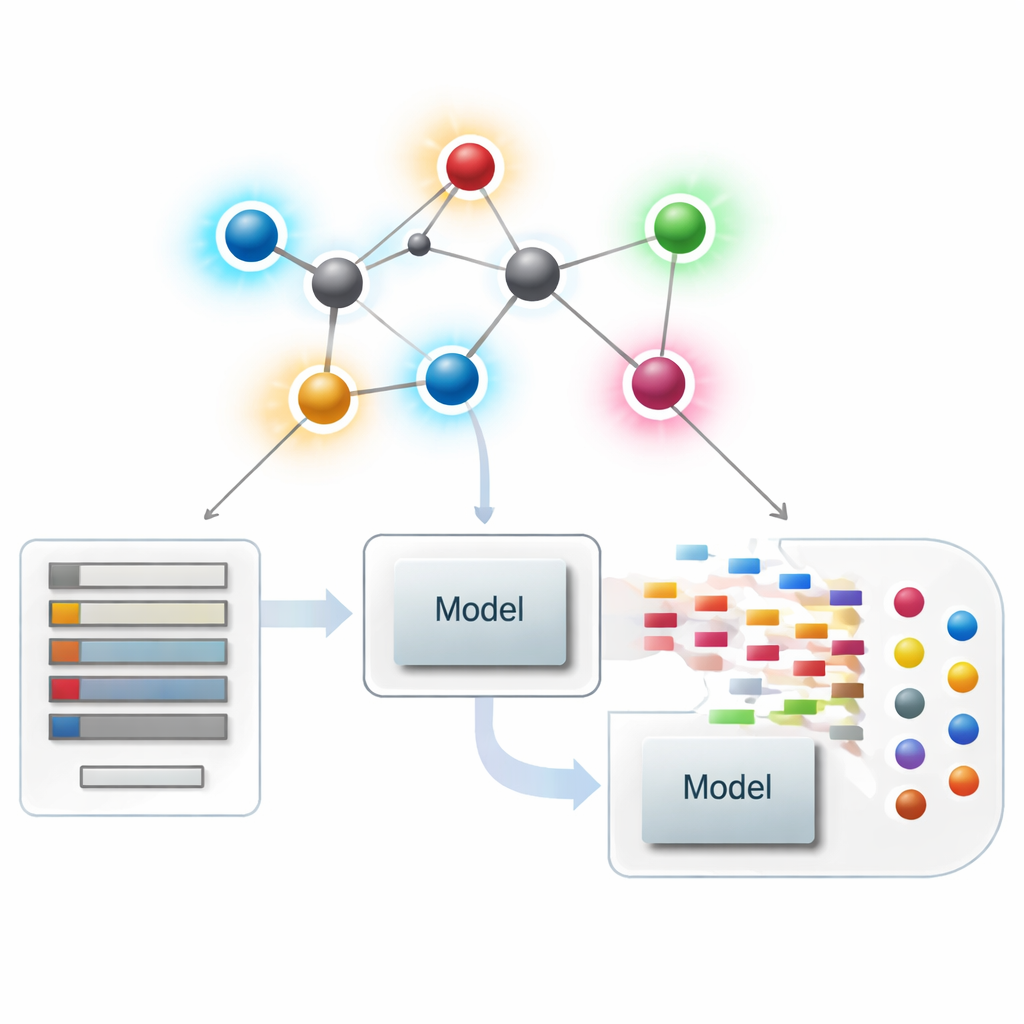
将分子视为网络
在现代化学信息学中,分子可以视为一个网络,每个原子是一个节点,每条化学键是一条连线。从这些网络中,科学家计算出数值汇总——称为指标或描述符——以捕捉分子的形状、分支和连通性等特征。传统描述符主要关注有多少键连接到每个原子,即所谓的度数。这些以原子为中心的汇总在将结构与沸点、溶解度或药物相容性等性质关联方面非常成功,但它们可能遗漏那些在整体上看起来相似却表现截然不同的分子之间的微妙差异。
把注意力放在化学键上
作者提出了一类新的“改良基于键的指标”,有意将关注点从原子转向化学键。对于分子网络中的每一条键,他们观察该键两端原子的度数,并将其组合成一个局部键因子,用来衡量该键邻域的拥挤程度。然后,这个因子对各种熟悉的基于度数的公式进行缩放。实际上,每条键获得一个分数,既反映其端点又反映其周围的拥塞程度。位于分子繁忙区域的键会被降权,而位于相对稀疏区域的键则稍微被放大,从而使总体描述符对局部重排(例如侧链不同排列)更为敏感。
在理想化网络上测试数学性质
在将这些新指标应用于真实分子之前,团队先在数学家熟悉的标准理想化网络族上分析它们:路径、环、完全图、星形图以及若干更复杂的“装置”结构。对于十六种改良的基于键的指标中的每一种,他们推导出紧凑的公式,说明当网络变得更大或更连通时指标如何增长。他们还证明了将指标值与一些基本特征(如最少和最多连接节点的连接数)关联的尖锐界。这些数学结果表明,新的以键为中心的描述符表现得可控且可预测,并且在非常规则的结构上通常简化为简单的重缩放,这有助于解释它们并将其与较旧指标进行比较。
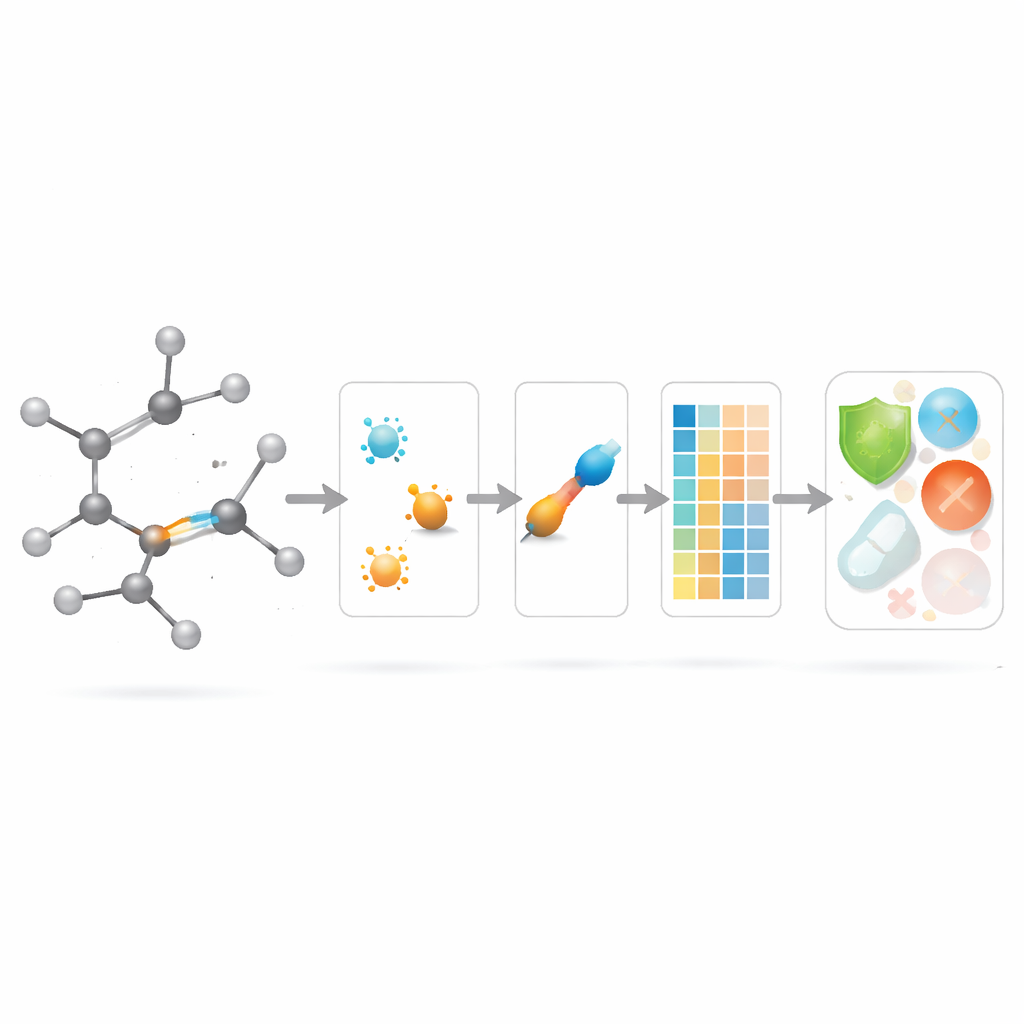
在药物建模中应用新的键评分
在理论到位之后,作者探问这些以键为中心的描述符在实践中是否确有帮助。他们从ChEMBL数据库中汇编了经整理的3,219个抗菌分子,并考虑十个连续目标:九个基本理化量(例如分子量、极性、表面积以及供、受氢键基团数等)加上一个抗菌强度的度量。随后他们构建了一个大的“模型动物园”回归方法,从简单线性拟合到现代树模型与提升算法,并比较三种情景:仅使用新的基于键的指标,仅使用标准理化性质,以及两者合用。
关于以键识别的描述符的结论
在所有十个目标上,常规模拟理化描述符给出了强有力的预测,这反映了这些度量经过数十年优化的结果。单独使用基于键的指标表现明显较差,表明它们不能完全取代标准特征。然而,当基于键的指标与理化描述符结合使用时,整体预测质量有所提升:跨目标的平均测试准确度略有增加,且一个无单位误差评分约下降了3%。在对结构敏感的量(如可旋转键数和“天然产物相似性”评分)上增益最为明显,在这些情况下详细的连通性显然重要。对于抗菌效力,所有模型仍然表现平平,表明要捕捉复杂生物活性仍需更丰富的信息。
给非专业读者的要点
这项研究表明,将化学键作为分子描述中的一等公民,可以为计算模型提供额外、有用的信息,尤其是与传统的整体化学性质结合时。新的以键为中心的描述符在数学上行为良好、易于计算,并有助于捕捉分子之间的微妙结构差异。尽管它们并不能单独解决药物发现问题,但它们提供了一层实用的新结构细节,能够在抗菌化合物的多属性建模中带来适度但持续的预测改进。
引用: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
关键词: 化学信息学, 分子描述符, 图论, QSAR QSPR, 抗菌药物发现