Clear Sky Science · ja
グラフ理論から化学情報学へ:修正結合ベース指標と仮説駆動型マルチタスクQSAR/QSPRベンチマーク
なぜ微小な分子のつながりが重要なのか
化学者はしばしば分子を小さな都市のように描写します:原子が建物で、結合が道路です。何十年にもわたり、分子の振る舞いを予測するための数学的手法の多くは「建物」で起きていること、つまり原子側に注目する傾向が強く、「道路」である結合自体にはあまり注目してきませんでした。本稿は単純だが強力な問いを投げかけます:結合そのものにもっと注意を払ったらどうなるか。その追加情報は、潜在的な抗菌薬の挙動をコンピュータがより良く予測するのに役立つだろうか?
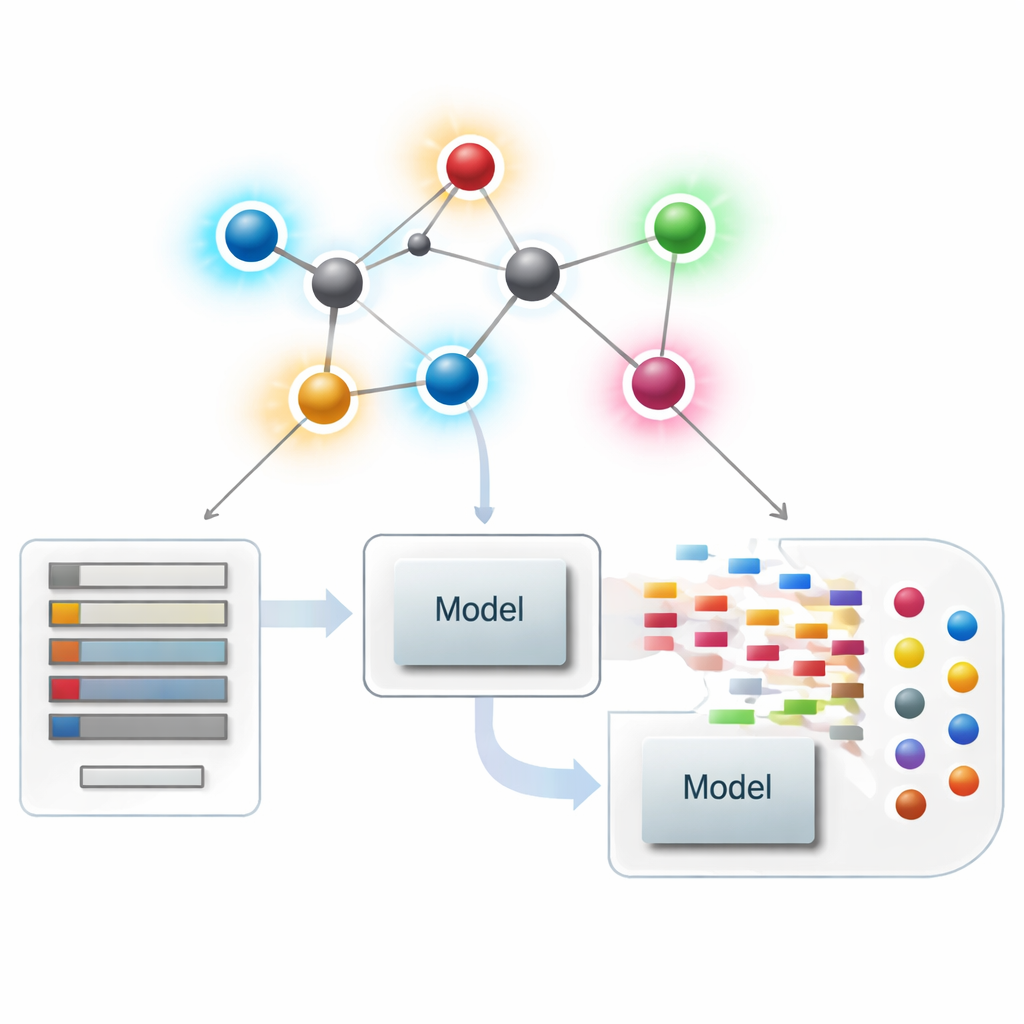
分子をネットワークとして見る
現代の化学情報学では、分子は各原子が点、各化学結合が線で表されるネットワークとして扱えます。こうしたネットワークから、分子の形状や分岐、結合性をとらえる数値的要約(指標や記述子と呼ばれる)を計算します。古典的な記述子は主に各原子に触れる結合の数、すなわち次数に焦点を当ててきました。原子中心のこれらの要約は沸点、溶解度、薬物らしさなどの性質と構造を結び付けるうえで非常に成功していますが、全体として似て見えるにもかかわらず非常に異なる振る舞いをする分子の微妙な違いを見落としがちです。
結合をスポットライトに置く
著者らは原子から結合へと意図的に注目を移す新しい一群の「修正結合ベース指標」を導入します。分子ネットワークの各結合について、それが接続する二つの原子の次数を見て、それらを組み合わせることで結合の周辺がどれだけ混雑しているかを測る局所的な結合係数を作ります。この係数は既存の次数ベースの式のさまざまなものをスケーリングします。実質的に各結合は両端の原子と周辺の混雑度を反映するスコアを与えられます。分子の混み合った領域の結合は重みが下がり、静かな領域の結合はやや重要度が上がるため、側鎖の異なる配列など局所的な再配置に対して記述子がより敏感になります。
理想化されたネットワークで数学を試す
これらの新しい指標を実際の分子で使う前に、研究チームは数学者に良く知られた標準的なネットワーク族(パス、サイクル、完全グラフ、スター、そしていくつかのより精巧な“ガジェット”構造)上で解析します。16種類の修正結合ベース指標それぞれについて、ネットワークが大きくなったりより結びつきが強くなったりするにつれて指標がどのように増大するかを示す簡潔な式を導きます。また、指標値を最小・最大接続ノードのような基本的特徴に結び付ける厳密な上界・下界を証明します。これらの数学的結果は、新しい結合重視の記述子が制御され予測可能な振る舞いを示すこと、非常に規則的な構造では単純な再スケーリングに帰着することが多く、そのため解釈や既存指標との比較が容易になることを示しています。
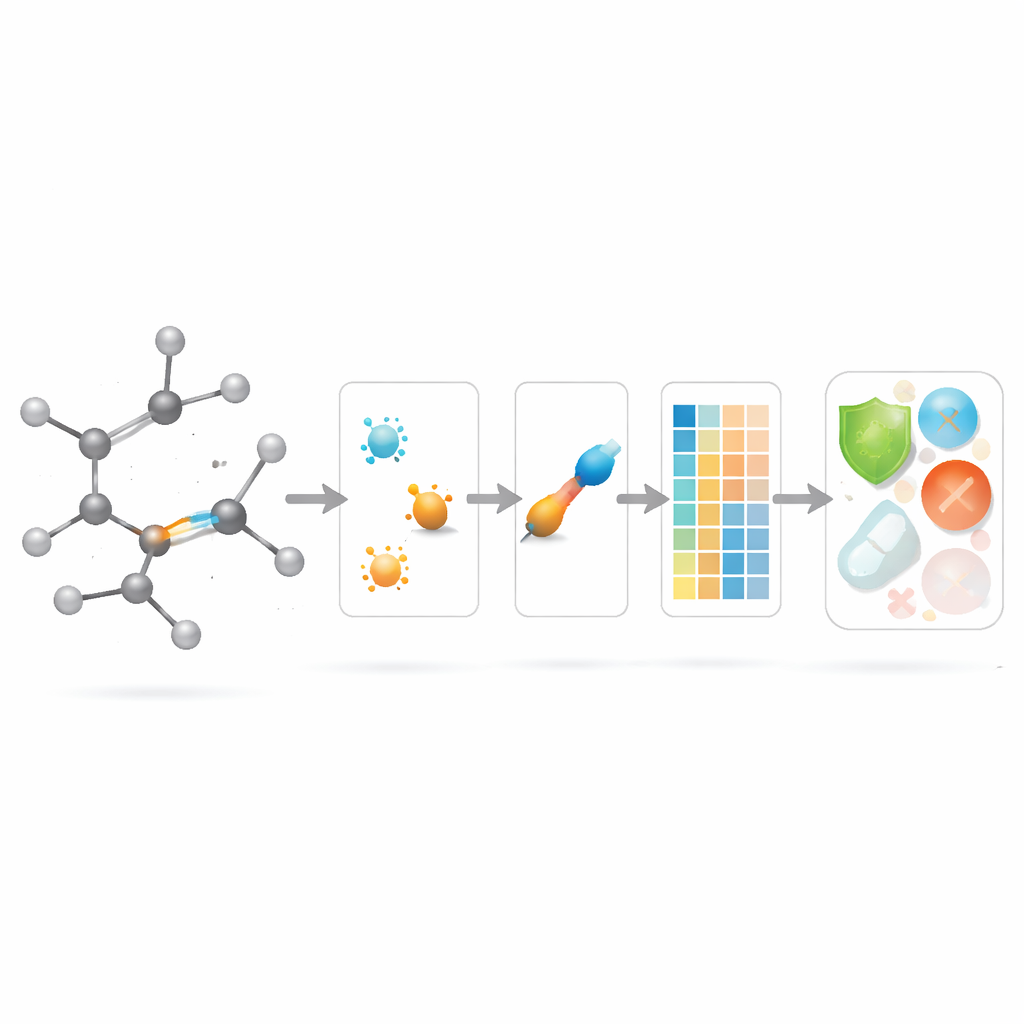
薬物モデリングで新しい結合スコアを活用する
理論が整った後、著者らはこれらの結合中心の記述子が実際に役立つかどうかを問います。彼らはChEMBLデータベースから厳選した3,219件の抗菌分子セットを組み、10の連続的ターゲットを考慮します:分子量、極性、表面積、供与体・受容体の水素結合数など9つの基本的物性量と、抗菌活性の指標です。そのうえで、単純な線形回帰から最新の木ベースやブースティング手法まで多数の回帰モデルを揃え、大規模な“モデル動物園”を構築し、三つのシナリオを比較します:新しい結合ベース指標のみ、標準的な物理化学的性質のみ、そして両者を併用した場合です。
結合認識型記述子に関する結果の示すところ
10のすべてのターゲットにわたって、従来の物理化学的記述子は堅牢な予測を示し、これらの指標が何十年にもわたって最適化されてきたことを反映しています。結合ベース指標単独では明らかに性能が劣り、従来の特徴の完全な代替にはならないことが示されました。しかし、結合ベース指標を物理化学的記述子と組み合わせると、総合的な予測精度が改善します:ターゲット全体での平均テスト精度がわずかに上がり、単位のない誤差スコアが約3パーセント低下しました。改善は可撓結合数や“天然物らしさ”スコアのような構造に敏感な量で最も顕著で、詳細な結合性が明らかに重要であることを示しています。抗菌活性についてはすべてのモデルが控えめな性能にとどまり、複雑な生物学的活性を捉えるにはさらに豊富な情報が必要であることが示唆されます。
専門外の読者へのまとめ
本研究は、化学結合を分子記述において第一級の要素として扱うことで、特に伝統的な総体的化学特性と組み合わせた場合に、コンピュータモデルに有用な付加情報を与えうることを示しています。新しい結合意識型指標は数学的に挙動が良好で計算が容易であり、分子間の微妙な構造差をとらえるのに役立ちます。それ単独で創薬を解決するわけではありませんが、抗菌化合物のマルチプロパティモデリングにおいて予測を控えめながらも一貫して改善する実用的な新たな構造情報の層を提供します。
引用: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
キーワード: 化学情報学, 分子記述子, グラフ理論, QSAR QSPR, 抗菌薬創薬