Clear Sky Science · es
De la teoría de grafos a la quimioinformática: índices modificados basados en enlaces y un banco de pruebas QSAR/QSPR multitarea impulsado por hipótesis
Por qué importan las pequeñas conexiones moleculares
Los químicos a menudo describen las moléculas como si fueran pequeñas ciudades: los átomos son los edificios y los enlaces son las carreteras. Durante décadas, la mayoría de las herramientas matemáticas para predecir el comportamiento de una molécula se han centrado en contar lo que ocurre en los “edificios” más que en las “carreteras” que los conectan. Este artículo plantea una pregunta simple pero potente: ¿y si prestamos más atención a los propios enlaces, y puede ese detalle extra ayudar a los ordenadores a predecir mejor cómo se comportarán posibles fármacos antibacterianos?
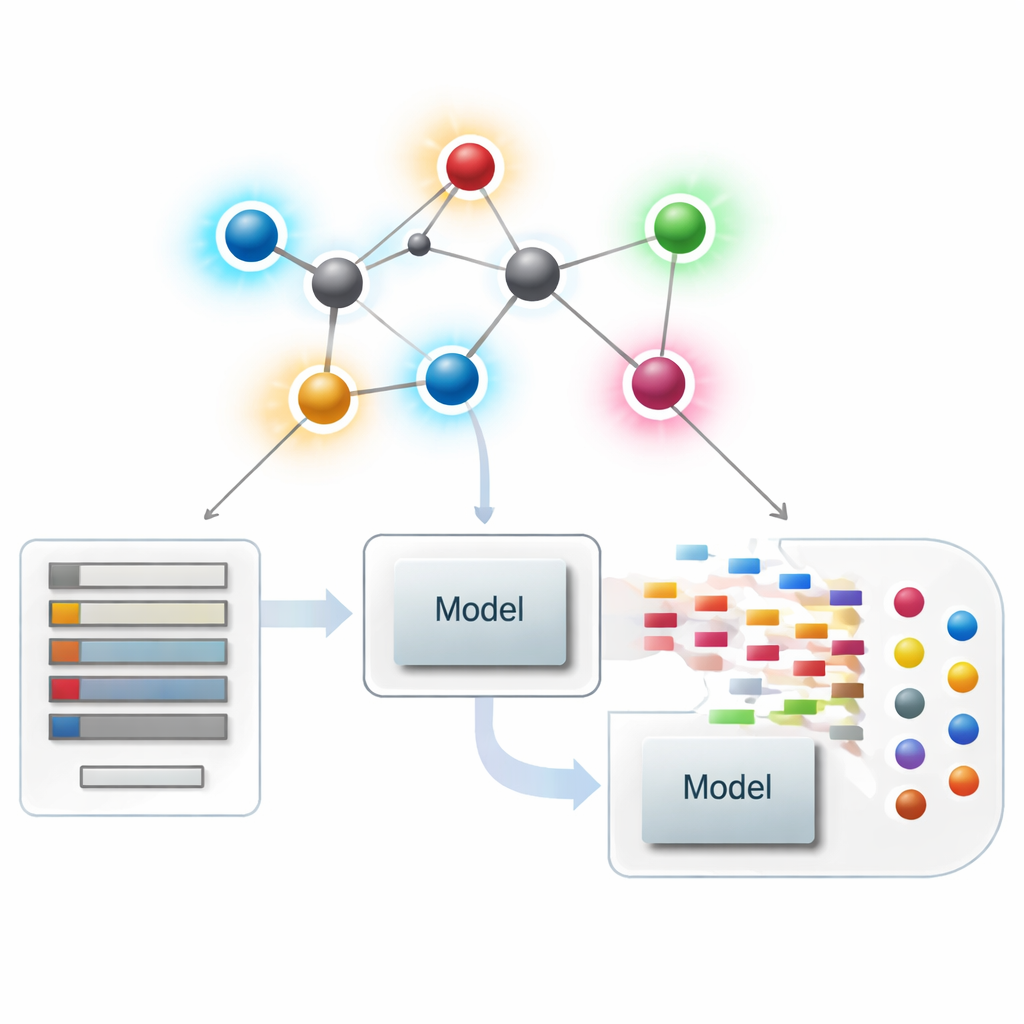
Ver las moléculas como redes
En la quimioinformática moderna, una molécula puede tratarse como una red, donde cada átomo es un punto y cada enlace químico es una línea. A partir de estas redes, los científicos calculan resúmenes numéricos —denominados índices o descriptores— que capturan aspectos de la forma molecular, la ramificación y la conectividad. Los descriptores clásicos se centran sobre todo en cuántos enlaces toca cada átomo, una magnitud llamada su grado. Estos resúmenes centrados en el átomo han tenido mucho éxito relacionando la estructura con propiedades como el punto de ebullición, la solubilidad o la semejanza a fármacos, pero pueden pasar por alto diferencias sutiles entre moléculas que parecen similares a escala global pero actúan de forma muy distinta.
Poner los enlaces en el centro
Los autores introducen una nueva familia de “índices modificados basados en enlaces” que desplazan deliberadamente la atención de los átomos hacia los enlaces. Para cada enlace en una red molecular, consideran los grados de los dos átomos que conecta y los combinan en un factor local del enlace que mide cuán congestionado está su vecindario. Este factor escala luego una variedad de fórmulas familiares basadas en grados. En efecto, cada enlace obtiene una puntuación que refleja tanto sus extremos como la congestión circundante. Los enlaces en regiones concurridas de una molécula se ponderan a la baja, mientras que los enlaces en regiones más tranquilas cuentan un poco más, haciendo que el descriptor global sea más sensible a reordenamientos locales como distintas disposiciones de cadenas laterales.
Probar las matemáticas en redes idealizadas
Antes de aplicar estos nuevos índices a moléculas reales, el equipo los analiza en familias estándar de redes idealizadas bien conocidas por los matemáticos: caminos, ciclos, grafos completos, estrellas y varias estructuras “gadget” más elaboradas. Para cada uno de los dieciséis índices modificados basados en enlaces, derivan fórmulas compactas que indican cómo crece el índice a medida que estas redes se hacen más grandes o más conectadas. También prueban cotas precisas que relacionan los valores del índice con características básicas como cuántas conexiones tienen los nodos menos y más conectados. Estos resultados matemáticos muestran que los nuevos descriptores centrados en enlaces se comportan de forma controlada y predecible y, a menudo, se reducen a simples reescalados en estructuras muy regulares, lo que ayuda a interpretarlos y a compararlos con índices anteriores.
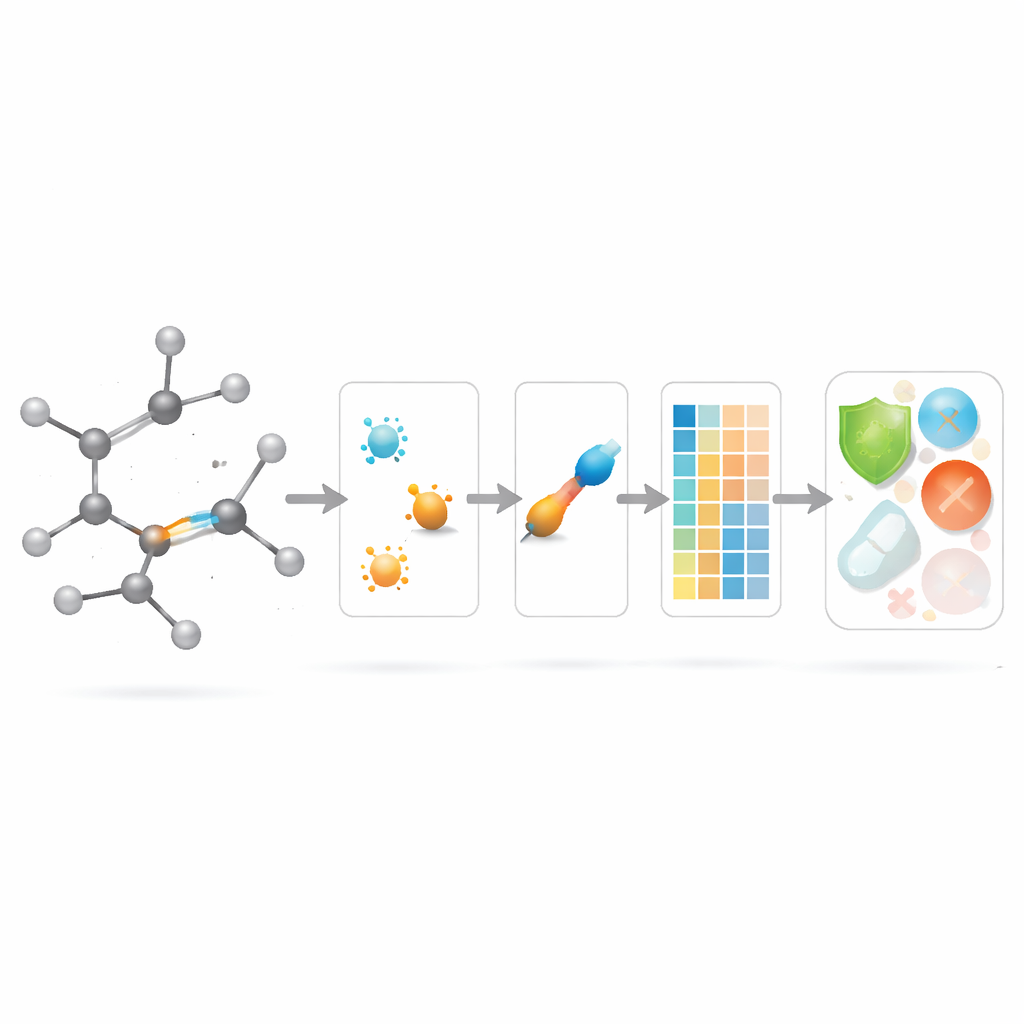
Aplicar las nuevas puntuaciones de enlaces en modelado farmacológico
Con la teoría establecida, los autores se preguntan si estos descriptores centrados en enlaces ayudan en la práctica. Reúnen un conjunto curado de 3.219 moléculas antibacterianas de la base de datos ChEMBL y consideran diez objetivos continuos: nueve magnitudes fisicoquímicas básicas (como peso molecular, polaridad, área superficial y conteos de donantes y aceptores de puente de hidrógeno) más una medida de potencia antibacteriana. A continuación construyen un gran “zoológico de modelos” de métodos de regresión, desde ajustes lineales sencillos hasta algoritmos modernos basados en árboles y boosting, y comparan tres escenarios: usar solo los nuevos índices basados en enlaces, usar solo propiedades fisicoquímicas estándar y usar ambas cosas combinadas.
Qué dicen los resultados sobre los descriptores conscientes de enlaces
En los diez objetivos, los descriptores fisicoquímicos habituales ofrecen predicciones sólidas, reflejando décadas de optimización de tales medidas. Los índices basados en enlaces por sí solos rinden notablemente peor, lo que muestra que no son un reemplazo completo de las características estándar. Sin embargo, cuando los índices basados en enlaces se combinan con descriptores fisicoquímicos, la calidad predictiva global mejora: la precisión media en prueba entre objetivos aumenta levemente y una puntuación de error adimensional disminuye alrededor de un tres por ciento. Las ganancias son más visibles para magnitudes sensibles a la estructura como el número de enlaces rotables y una puntuación de “semejanza a productos naturales”, donde la conectividad detallada importa claramente. Para la potencia antibacteriana, todos los modelos siguen siendo modestos, lo que sugiere que se necesita información aún más rica para capturar la actividad biológica compleja.
Mensaje clave para no especialistas
Este estudio muestra que tratar los enlaces químicos como ciudadanos de primera clase en las descripciones moleculares puede aportar información extra y útil para los modelos informáticos, especialmente cuando se combinan con propiedades químicas tradicionales y de amplio alcance. Los nuevos índices conscientes de enlaces son matemáticamente bien comportados, fáciles de calcular y ayudan a capturar diferencias estructurales sutiles entre moléculas. Aunque no resuelven por sí solos el descubrimiento de fármacos, ofrecen una nueva capa práctica de detalle estructural que puede mejorar de forma modesta pero consistente las predicciones en el modelado multitarea de propiedades de compuestos antibacterianos.
Cita: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Palabras clave: quimioinformática, descriptores moleculares, teoría de grafos, QSAR QSPR, descubrimiento de antibacterianos