Clear Sky Science · nl
Van grafentheorie naar chemoinformatica: aangepaste binding-gebaseerde indices en een hypothese-gedreven multi-task QSAR/QSPR benchmark
Waarom kleine moleculaire verbindingen ertoe doen
Chemici beschrijven moleculen vaak alsof het kleine stadjes zijn: atomen zijn de gebouwen en bindingen de wegen. Decennialang richtten de meeste wiskundige hulpmiddelen om het gedrag van een molecuul te voorspellen zich op wat er bij de "gebouwen" gebeurt in plaats van op de "wegen" ertussen. Dit artikel stelt een eenvoudige maar krachtige vraag: wat als we meer aandacht besteden aan de bindingen zelf, en kan die extra informatie computers helpen beter te voorspellen hoe potentiële antibacteriële geneesmiddelen zich zullen gedragen?
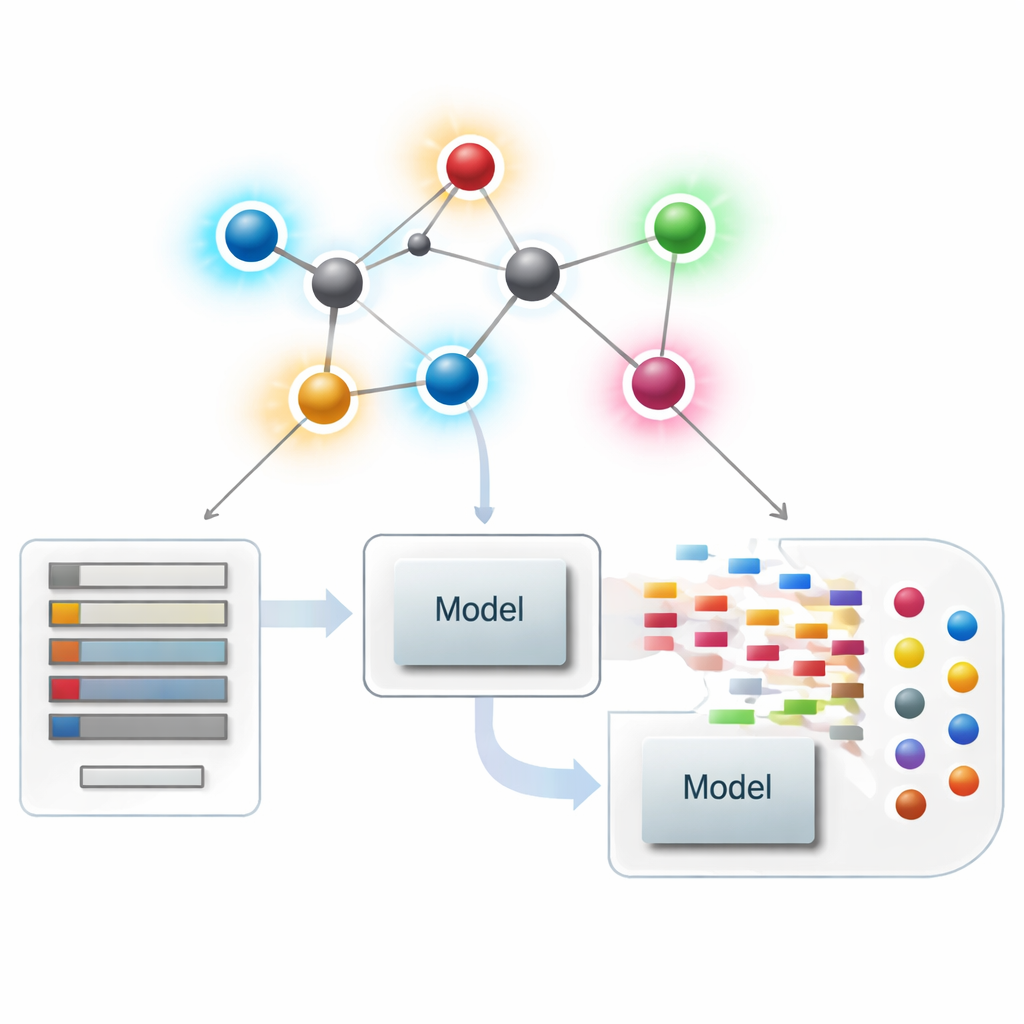
Moleculen bekijken als netwerken
In de moderne chemoinformatica kan een molecuul worden behandeld als een netwerk, waarbij elk atoom een knooppunt is en elke chemische binding een lijn. Uit deze netwerken berekenen wetenschappers numerieke samenvattingen — indices of descriptors genoemd — die aspecten van moleculaire vorm, vertakking en connectiviteit vastleggen. Klassieke descriptors richten zich vooral op hoeveel bindingen elk atoom raakt, een grootheid die zijn graad wordt genoemd. Deze atoomgerichte samenvattingen zijn zeer succesvol geweest in het relateren van structuur aan eigenschappen zoals kookpunt, oplosbaarheid of drug‑likeness, maar ze kunnen subtiele verschillen missen tussen moleculen die globaal vergelijkbaar lijken maar heel verschillend werken.
Bindingen in de schijnwerpers zetten
De auteurs introduceren een nieuwe familie van "aangepaste binding‑gebaseerde indices" die bewust de aandacht verschuiven van atomen naar bindingen. Voor elke binding in een moleculair netwerk bekijken ze de graden van de twee atomen die die binding verbinden en combineren die tot een lokale bindingsfactor die meet hoe druk de omgeving van die binding is. Deze factor schaalt vervolgens verschillende bekende graadgebaseerde formules. Effectief krijgt elke binding een score die zowel zijn eindpunten als de omliggende congestie weerspiegelt. Bindingen in drukke regio's van een molecuul worden minder zwaar gewogen, terwijl bindingen in rustigere regio's iets zwaarder tellen, waardoor de algemene descriptor gevoeliger wordt voor lokale herschikkingen zoals verschillende ordeningen van zijketens.
De wiskunde testen op geïdealiseerde netwerken
Voordat ze deze nieuwe indices op echte moleculen toepassen, analyseren de onderzoekers ze op standaardfamilies van geïdealiseerde netwerken die wiskundigen goed kennen: paden, cycli, volledige grafen, sterren en meerdere meer uitgewerkte "gadget"-structuren. Voor elk van de zestien aangepaste binding‑gebaseerde indices leiden ze compacte formules af die aangeven hoe de index groeit naarmate deze netwerken groter of meer verbonden worden. Ze bewijzen ook scherpe grenzen die indexwaarden relateren aan basiskenmerken zoals hoeveel verbindingen de minst en meest verbonden knopen hebben. Deze wiskundige resultaten tonen aan dat de nieuwe op bindingen gerichte descriptors zich gecontroleerd en voorspelbaar gedragen en vaak neerkomen op eenvoudige herschaling in zeer regelmatige structuren, wat helpt bij de interpretatie en vergelijking met oudere indices.
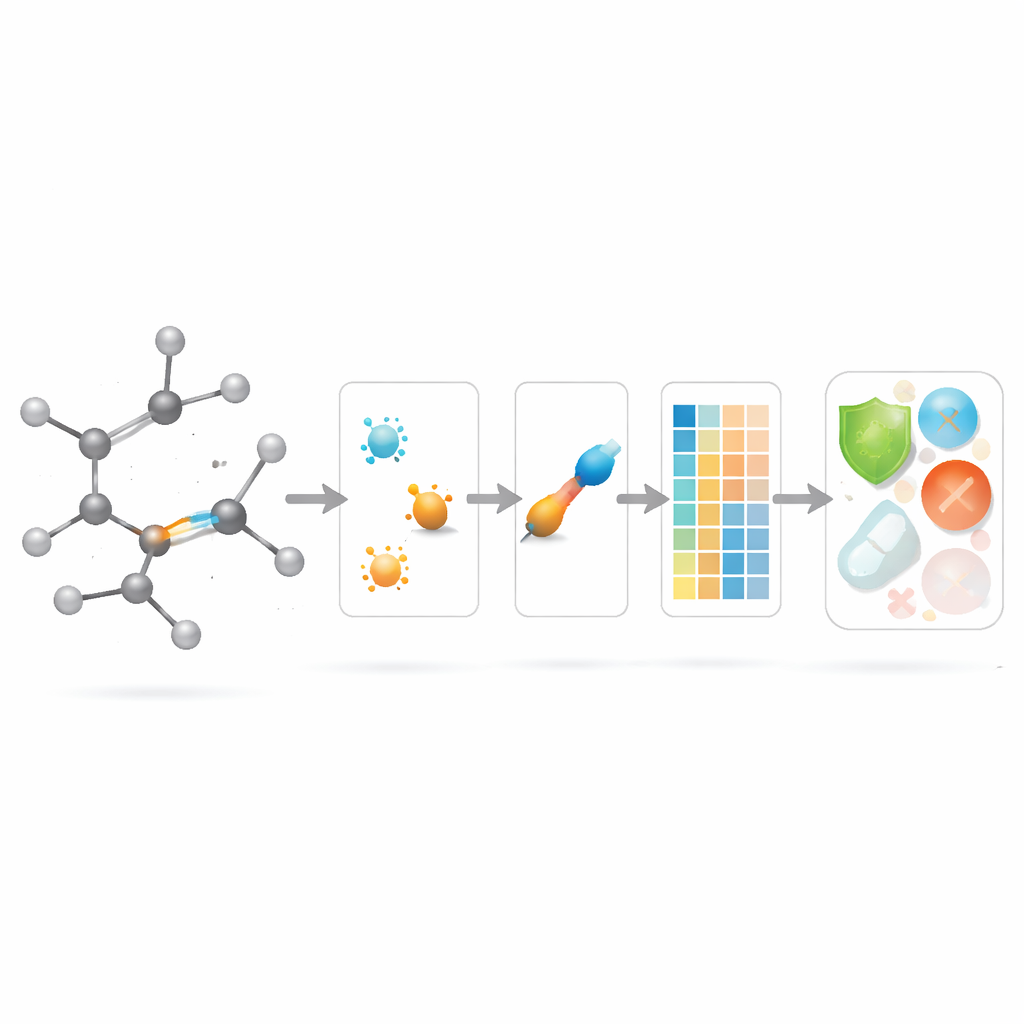
De nieuwe binding-scores inzetten in geneesmiddelmodellering
Met de theoretische basis op zijn plaats vragen de auteurs of deze op bindingen gerichte descriptors in de praktijk daadwerkelijk helpen. Ze stellen een gecureerde set samen van 3.219 antibacteriële moleculen uit de ChEMBL‑database en beschouwen tien continue doelen: negen basale fysisch‑chemische grootheden (zoals molecuulgewicht, polariteit, oppervlak en aantallen waterstof‑brugdonoren en -acceptoren) plus een maat voor antibacteriële sterkte. Vervolgens bouwen ze een omvangrijke "modelzoo" van regressiemethoden, van eenvoudige lineaire fits tot moderne boomgebaseerde en boosting‑algoritmen, en vergelijken drie scenario’s: alleen de nieuwe binding‑gebaseerde indices gebruiken, alleen standaard fysisch‑chemische eigenschappen gebruiken, en beiden samen gebruiken.
Wat de resultaten zeggen over binding‑bewuste descriptors
Over alle tien doelen geven de gebruikelijke fysisch‑chemische descriptors sterke voorspellingen, wat de decennialange optimalisatie van zulke grootheden weerspiegelt. De binding‑gebaseerde indices presteren op zichzelf merkbaar minder goed, wat aantoont dat ze geen volledige vervanging zijn voor standaardkenmerken. Wanneer de binding‑gebaseerde indices echter worden gecombineerd met fysisch‑chemische descriptors, verbetert de algemene voorspellingskwaliteit: de gemiddelde testnauwkeurigheid over doelen neemt licht toe en een eenheidsvrije foutscore daalt met ongeveer drie procent. De winst is het meest zichtbaar voor structuur‑gevoelige grootheden zoals het aantal roteerbare bindingen en een score voor "natural product‑likeness", waar gedetailleerde connectiviteit duidelijk van belang is. Voor antibacteriële potentie blijven alle modellen bescheiden, wat suggereert dat nog rijkere informatie nodig is om complexe biologische activiteit vast te leggen.
Belangrijkste boodschap voor niet‑specialisten
Deze studie toont aan dat het behandelen van chemische bindingen als volwaardige elementen in moleculaire beschrijvingen extra, bruikbare informatie kan opleveren voor computermodellen, vooral wanneer ze worden gecombineerd met traditionele, bulk chemische eigenschappen. De nieuwe binding‑bewuste indices zijn wiskundig goed gedisciplineerd, gemakkelijk te berekenen en helpen subtiele structurele verschillen tussen moleculen vast te leggen. Hoewel ze de geneesmiddelontdekking niet op zichzelf oplossen, bieden ze een praktische nieuwe laag van structurele detaillering die bescheiden maar consistent voorspellingen kan verbeteren in multi‑eigenschapsmodellering van antibacteriële verbindingen.
Bronvermelding: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Trefwoorden: chemoinformatica, moleculaire descriptors, grafentheorie, QSAR QSPR, ontdekking van antibacteriële geneesmiddelen