Clear Sky Science · en
From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark
Why tiny molecular connections matter
Chemists often describe molecules as if they were tiny cities: atoms are the buildings and bonds are the roads. For decades, most mathematical tools for predicting how a molecule behaves have focused on counting what happens at the “buildings” rather than on the “roads” between them. This paper asks a simple but powerful question: what if we pay closer attention to the bonds themselves, and can that extra detail help computers better predict how potential antibacterial drugs will behave?
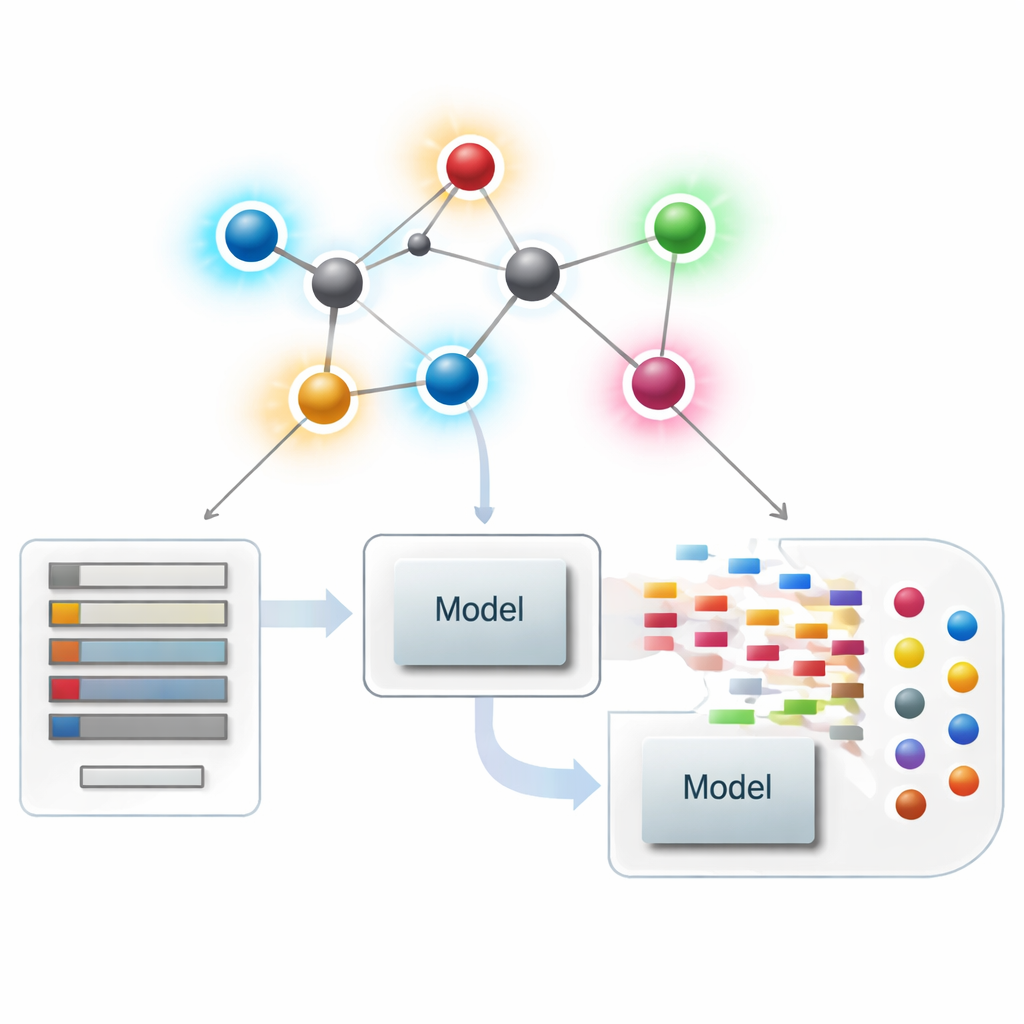
Looking at molecules as networks
In modern chemoinformatics, a molecule can be treated as a network, where each atom is a point and each chemical bond is a line. From these networks, scientists compute numerical summaries—called indices or descriptors—that capture aspects of molecular shape, branching, and connectivity. Classic descriptors mostly focus on how many bonds touch each atom, a quantity called its degree. These atom-centered summaries have been very successful in relating structure to properties such as boiling point, solubility, or drug-likeness, but they can miss subtle differences between molecules that look globally similar yet act very differently.
Putting bonds in the spotlight
The authors introduce a new family of “modified bond-based indices” that deliberately shift attention from atoms to bonds. For every bond in a molecular network, they look at the degrees of the two atoms it connects and combine them into a local bond factor that measures how crowded the bond’s neighborhood is. This factor then scales a variety of familiar degree-based formulas. In effect, each bond gets a score that reflects both its endpoints and its surrounding congestion. Bonds in busy regions of a molecule are down‑weighted, while bonds in quieter regions count a bit more, making the overall descriptor more sensitive to local rearrangements such as different arrangements of side chains.
Testing the mathematics on idealized networks
Before using these new indices on real molecules, the team analyzes them on standard families of idealized networks that mathematicians know well: paths, cycles, complete graphs, stars, and several more elaborate “gadget” structures. For each of sixteen modified bond-based indices, they derive compact formulas that tell how the index grows as these networks get larger or more connected. They also prove sharp bounds that relate index values to basic features such as how many connections the least- and most‑connected nodes have. These mathematical results show that the new bond-focused descriptors behave in a controlled, predictable way and often reduce to simple rescalings on very regular structures, which helps interpret them and compare them to older indices.
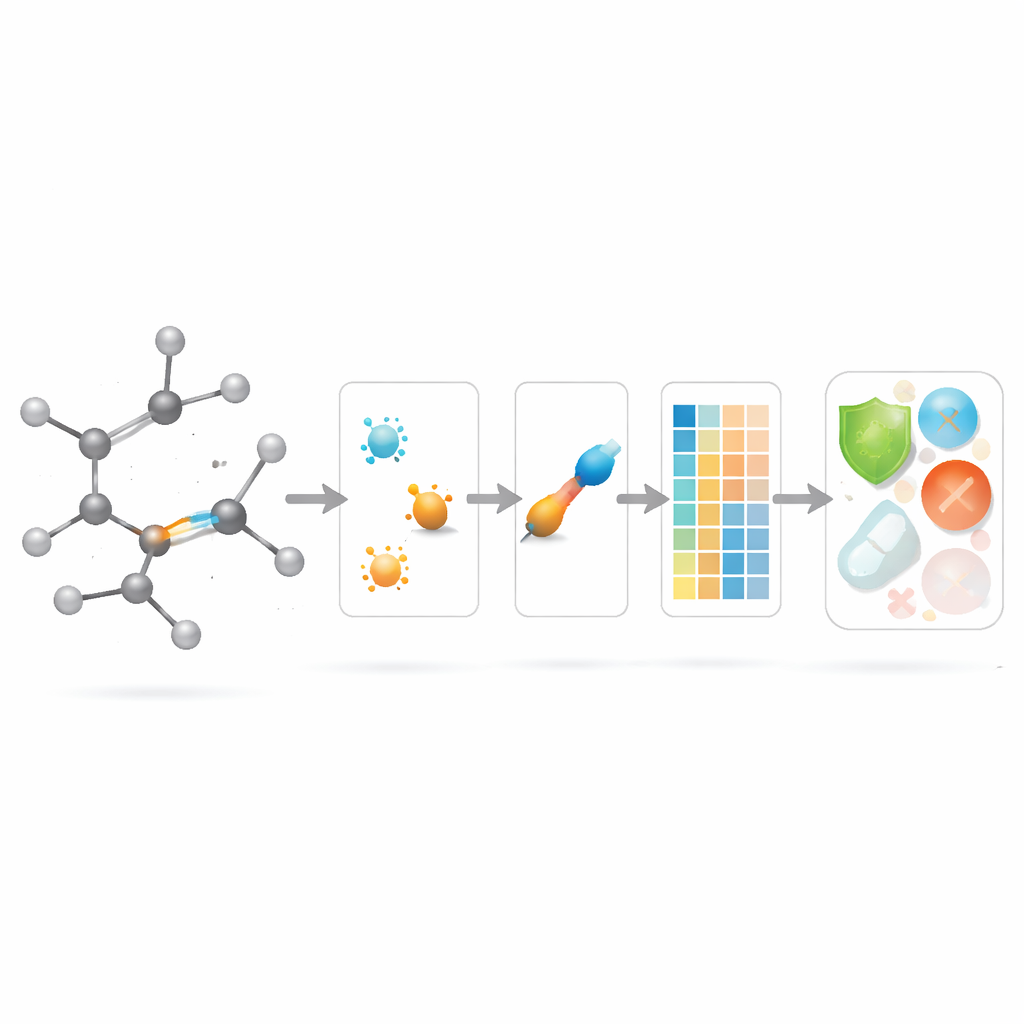
Putting new bond scores to work in drug modelling
With the theory in place, the authors ask whether these bond-centric descriptors actually help in practice. They assemble a curated set of 3,219 antibacterial molecules from the ChEMBL database and consider ten continuous targets: nine basic physicochemical quantities (such as molecular weight, polarity, surface area, and counts of hydrogen‑bond donors and acceptors) plus a measure of antibacterial strength. They then build a large “model zoo” of regression methods, from simple linear fits to modern tree‑based and boosting algorithms, and compare three scenarios: using only the new bond-based indices, using only standard physicochemical properties, and using both together.
What the results say about bond-aware descriptors
Across all ten targets, the usual physicochemical descriptors give strong predictions, reflecting decades of optimization of such measures. The bond-based indices by themselves perform noticeably worse, showing that they are not a complete replacement for standard features. However, when the bond-based indices are combined with physicochemical descriptors, overall prediction quality improves: the average test accuracy across targets increases slightly, and a unit‑free error score decreases by about three percent. The gains are most visible for structure‑sensitive quantities like the number of rotatable bonds and a “natural product‑likeness” score, where detailed connectivity clearly matters. For antibacterial potency, all models remain modest, suggesting that even richer information is needed to capture complex biological activity.
Take‑home message for non‑specialists
This study shows that treating chemical bonds as first‑class citizens in molecular descriptions can provide extra, useful information for computer models, especially when blended with traditional, bulk chemical properties. The new bond‑aware indices are mathematically well‑behaved, easy to compute, and help capture subtle structural differences between molecules. While they do not solve drug discovery on their own, they offer a practical new layer of structural detail that can modestly but consistently improve predictions in multi‑property modelling of antibacterial compounds.
Citation: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Keywords: chemoinformatics, molecular descriptors, graph theory, QSAR QSPR, antibacterial drug discovery