Clear Sky Science · sv
Från grafteori till kemoinformatik: modifierade bindningsbaserade index och en hypotesdriven multi‑task QSAR/QSPR‑benchmark
Varför små molekylära förbindelser spelar roll
Kemister beskriver ofta molekyler som om de vore små städer: atomerna är byggnaderna och bindningarna vägarna. Under årtionden har de flesta matematiska verktyg för att förutsäga hur en molekyl beter sig fokuserat på att räkna vad som händer vid ”byggnaderna” snarare än på ”vägarna” däremellan. Denna artikel ställer en enkel men kraftfull fråga: vad händer om vi lägger större vikt vid själva bindningarna — och kan den extra detaljen hjälpa datorer att bättre förutsäga hur potentiella antibakteriella läkemedel kommer att uppföra sig?
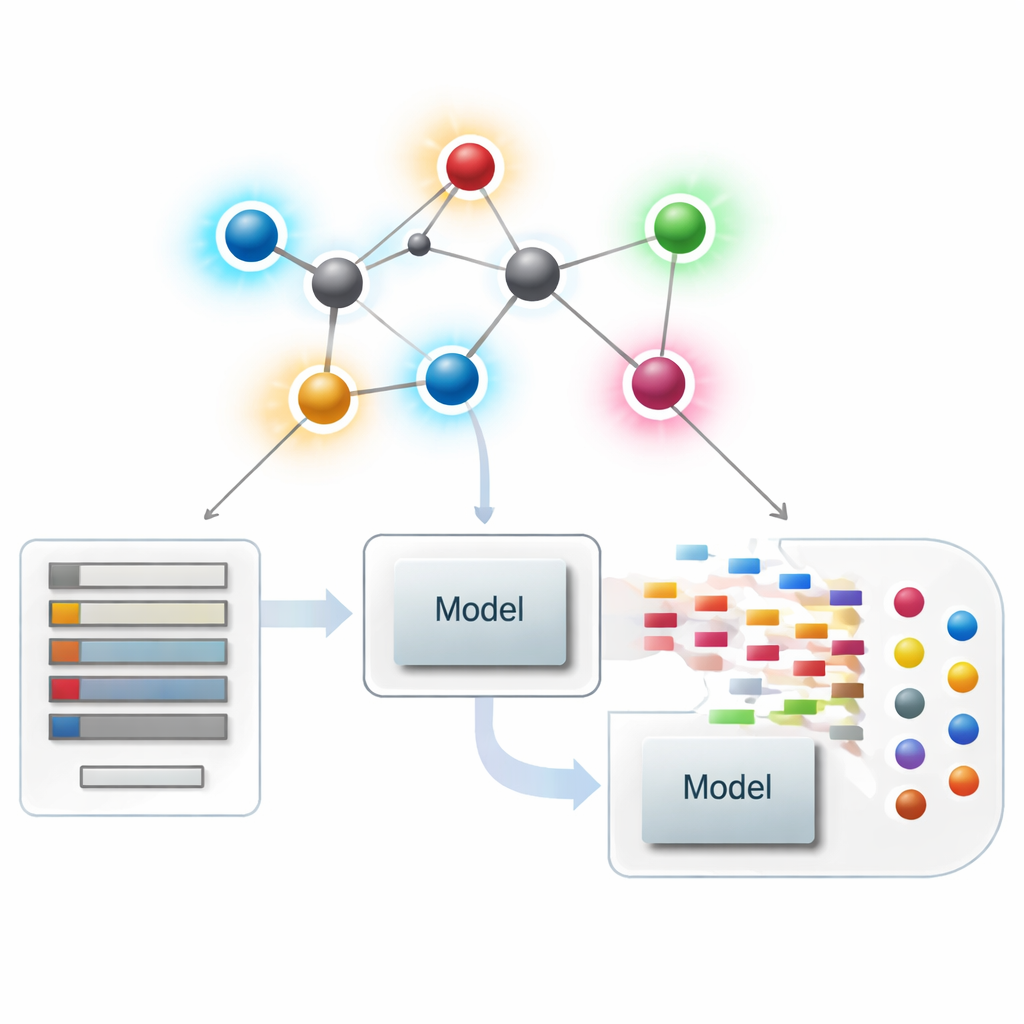
Att betrakta molekyler som nätverk
I modern kemoinformatik kan en molekyl behandlas som ett nätverk där varje atom är en punkt och varje kemisk bindning är en linje. Från dessa nätverk beräknar forskare numeriska sammanfattningar — kallade index eller deskriptorer — som fångar aspekter av molekylens form, förgrening och konnektivitet. Klassiska deskriptorer fokuserar mest på hur många bindningar som möter varje atom, en storhet som kallas dess grad. Dessa atomcentrerade sammanfattningar har varit mycket framgångsrika för att koppla struktur till egenskaper som kokpunkt, löslighet eller läkemedelslikhet, men de kan missa subtila skillnader mellan molekyler som ser globellt lika ut men beter sig väldigt olika.
Sätter bindningarna i rampljuset
Författarna introducerar en ny familj av "modifierade bindningsbaserade index" som avsiktligt flyttar uppmärksamheten från atomer till bindningar. För varje bindning i ett molekylärt nätverk betraktar de graderna hos de två atomerna den förbinder och kombinerar dem till en lokal bindningsfaktor som mäter hur trångt det är i bindningens omgivning. Denna faktor skalar sedan en rad välkända gradbaserade formler. I praktiken får varje bindning en poäng som återspeglar både dess ändpunkter och dess omgivande trängsel. Bindningar i upptagna regioner av en molekyl nedvägs medan bindningar i lugnare områden räknas lite mer, vilket gör den övergripande deskriptorn mer känslig för lokala omarrangemang som olika arrangemang av sidokedjor.
Testar matematiken på idealiserade nätverk
Innan dessa nya index används på verkliga molekyler analyserar teamet dem på standardfamiljer av idealiserade nätverk som matematiker känner väl: stigar, cykler, fullständiga grafer, stjärnor och flera mer invecklade "gadget"‑strukturer. För var och en av sexton modifierade bindningsbaserade index härleder de kompakta formler som visar hur indexet växer när dessa nätverk blir större eller mer sammankopplade. De bevisar också skarpa gränser som relaterar indexvärden till grundläggande egenskaper som hur många förbindelser de minst och mest förbundna noderna har. Dessa matematiska resultat visar att de nya bindningsfokuserade deskriptorerna beter sig på ett kontrollerat, förutsägbart sätt och ofta reduceras till enkla omskalningar på mycket regelbundna strukturer, vilket underlättar tolkning och jämförelse med äldre index.
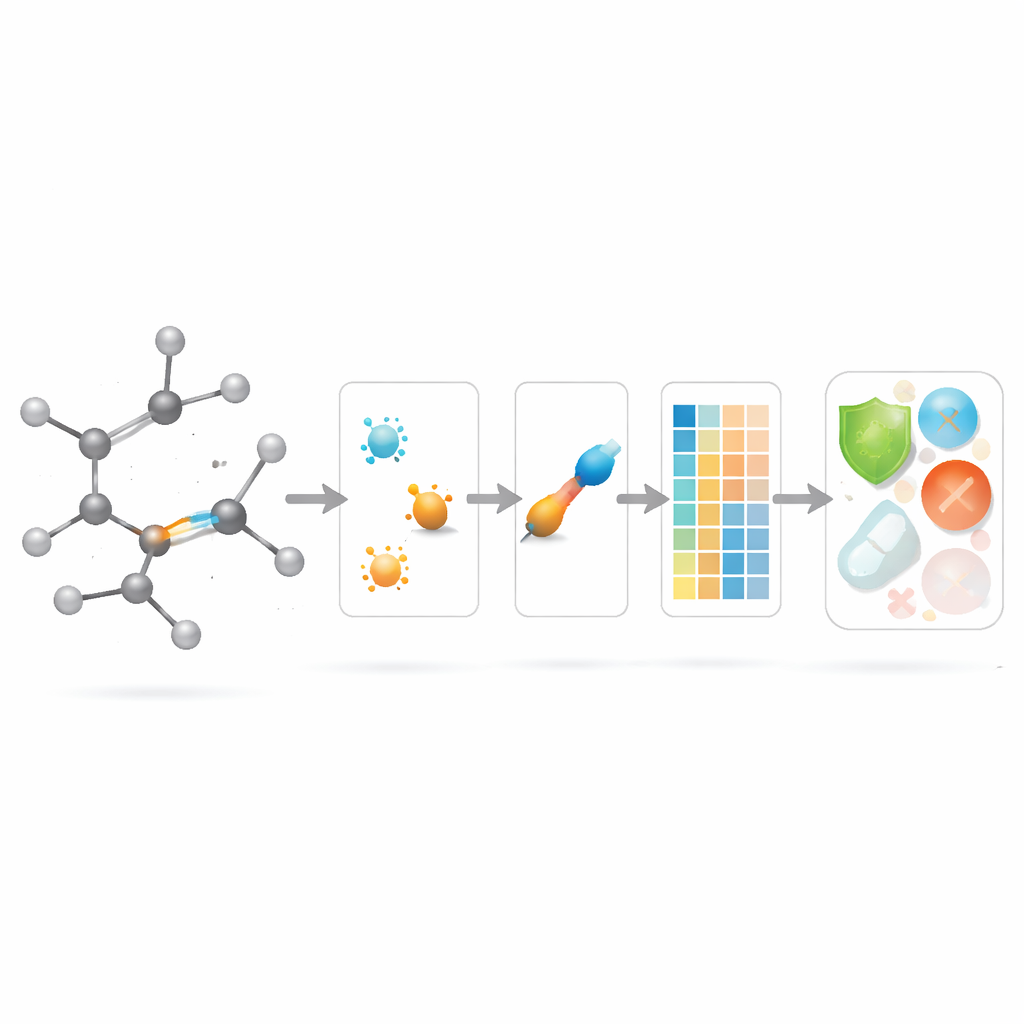
Använder nya bindningspoäng i läkemedelsmodellering
Med teorin på plats frågar författarna om dessa bindningscentrerade deskriptorer faktiskt hjälper i praktiken. De sammanställer en kurerad uppsättning om 3 219 antibakteriella molekyler från ChEMBL‑databasen och betraktar tio kontinuerliga mål: nio grundläggande fysikokemiska storheter (såsom molekylvikt, polaritet, yta och antal vätebindningsdonatorer och -acceptorer) plus ett mått på antibakteriell styrka. De bygger sedan en stor "modellzoo" av regressionsmetoder, från enkla linjära anpassningar till moderna träd‑ och boostingalgoritmer, och jämför tre scenarier: att använda endast de nya bindningsbaserade indexen, att använda endast standard fysikokemiska egenskaper, och att använda båda tillsammans.
Vad resultaten säger om bindningsmedvetna deskriptorer
Över alla tio mål ger de vanliga fysikokemiska deskriptorerna starka prediktioner, vilket speglar årtionden av optimering av sådana mått. De bindningsbaserade indexen presterar avsevärt sämre på egen hand, vilket visar att de inte är en fullständig ersättning för standardfunktioner. När de bindningsbaserade indexen kombineras med fysikokemiska deskriptorer förbättras dock den övergripande prediktionskvaliteten: den genomsnittliga testnoggrannheten över målen ökar något och ett enhetsfritt felmått minskar med ungefär tre procent. Vinsterna är mest synliga för strukturkänsliga storheter som antalet roterbara bindningar och ett mått på "naturlig produkt‑likhet", där detaljerad konnektivitet tydligt spelar roll. För antibakteriell potens förblir alla modeller blygsamma, vilket tyder på att ännu rikare information behövs för att fånga komplex biologisk aktivitet.
Huvudbudskap för icke‑specialister
Denna studie visar att att betrakta kemiska bindningar som förstklassiga medborgare i molekylära beskrivningar kan ge extra, användbar information för datorbaserade modeller, särskilt när de blandas med traditionella, övergripande kemiska egenskaper. De nya bindningsmedvetna indexen är matematiskt välbeteende, lätta att beräkna och hjälper till att fånga subtila strukturella skillnader mellan molekyler. Även om de inte löser läkemedelsupptäckt på egen hand erbjuder de ett praktiskt nytt lager av strukturell detalj som kan förbättra prediktionerna i multi‑egenskapsmodellering av antibakteriella föreningar, om än måttligt men konsekvent.
Citering: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Nyckelord: kemoinformatik, molekylära deskriptorer, grafteori, QSAR QSPR, upptäckt av antibakteriella läkemedel