Clear Sky Science · fr
De la théorie des graphes à la chimiinformatique : indices modifiés basés sur les liaisons et un banc d’essai multi‑tâches QSAR/QSPR guidé par des hypothèses
Pourquoi les petites liaisons moléculaires comptent
Les chimistes décrivent souvent les molécules comme de petites cités : les atomes sont les bâtiments et les liaisons sont les routes. Pendant des décennies, la plupart des outils mathématiques visant à prédire le comportement d’une molécule se sont concentrés sur ce qui se passe aux « bâtiments » plutôt que sur les « routes » qui les relient. Cet article pose une question simple mais puissante : et si l’on accordait davantage d’attention aux liaisons elles‑mêmes, et ce supplément de détail pourrait‑il aider les ordinateurs à mieux prédire le comportement de molécules candidates en tant qu’antibiotiques ?
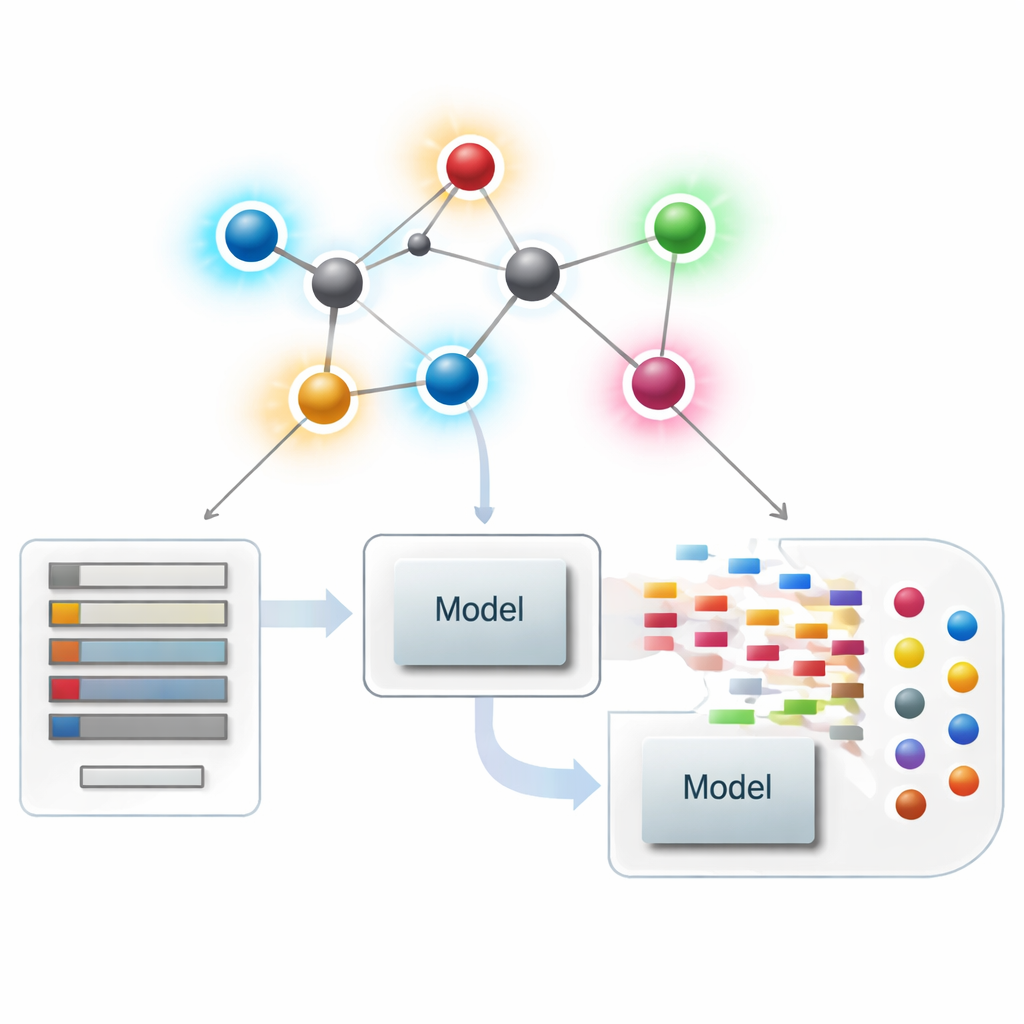
Considérer les molécules comme des réseaux
En chimiinformatique moderne, une molécule peut être traitée comme un réseau, où chaque atome est un point et chaque liaison chimique une arête. À partir de ces réseaux, les chercheurs calculent des résumés numériques — appelés indices ou descripteurs — qui capturent des aspects de la forme moléculaire, du ramification et de la connectivité. Les descripteurs classiques se concentrent principalement sur le nombre de liaisons incidentes à chaque atome, une quantité appelée degré. Ces résumés centrés sur l’atome ont été très efficaces pour relier la structure à des propriétés telles que le point d’ébullition, la solubilité ou la conformité aux règles de drug‑likeness, mais ils peuvent manquer de différences subtiles entre des molécules qui se ressemblent globalement et agissent pourtant très différemment.
Mettre les liaisons sous les projecteurs
Les auteurs présentent une nouvelle famille d’« indices modifiés basés sur les liaisons » qui déplacent délibérément l’attention des atomes vers les liaisons. Pour chaque liaison dans un réseau moléculaire, ils examinent les degrés des deux atomes qu’elle relie et les combinent en un facteur local de liaison qui mesure l’encombrement du voisinage de la liaison. Ce facteur pondère ensuite diverses formules connues basées sur les degrés. En pratique, chaque liaison reçoit un score reflétant à la fois ses extrémités et la congestion environnante. Les liaisons situées dans des régions denses d’une molécule sont atténuées, tandis que celles dans des zones plus calmes pèsent un peu plus, rendant le descripteur global plus sensible aux réarrangements locaux tels que des dispositions différentes de chaînes latérales.
Tester les mathématiques sur des réseaux idéalisés
Avant d’appliquer ces nouveaux indices à des molécules réelles, l’équipe les analyse sur des familles standard de réseaux idéalisés bien connus des mathématiciens : chemins, cycles, graphes complets, étoiles et plusieurs structures « gadget » plus élaborées. Pour chacun des seize indices modifiés basés sur les liaisons, ils établissent des formules compactes qui décrivent comment l’indice évolue à mesure que ces réseaux deviennent plus grands ou plus connectés. Ils démontrent également des bornes nettes reliant les valeurs des indices à des caractéristiques de base telles que le nombre de connexions des nœuds les moins et les plus connectés. Ces résultats mathématiques montrent que les nouveaux descripteurs focalisés sur les liaisons se comportent de manière contrôlée et prévisible et se réduisent souvent à de simples mises à l’échelle sur des structures très régulières, ce qui facilite leur interprétation et leur comparaison aux indices plus anciens.
Mettre les nouveaux scores de liaison au service de la modélisation de médicaments
Avec la théorie en place, les auteurs se demandent si ces descripteurs centrés sur les liaisons sont réellement utiles en pratique. Ils assemblent un jeu de données soigné de 3 219 molécules antibactériennes issues de la base ChEMBL et considèrent dix cibles continues : neuf grandeurs physico‑chimiques de base (telles que masse moléculaire, polarité, surface, et nombres de donneurs et accepteurs de liaisons hydrogène) ainsi qu’une mesure de la puissance antibactérienne. Ils construisent ensuite un vaste « zoo » de modèles de régression, allant d’ajustements linéaires simples à des algorithmes modernes basés sur les arbres et le boosting, et comparent trois scénarios : n’utiliser que les nouveaux indices basés sur les liaisons, n’utiliser que les propriétés physico‑chimiques standard, et utiliser les deux ensemble.
Ce que disent les résultats sur les descripteurs sensibles aux liaisons
Sur l’ensemble des dix cibles, les descripteurs physico‑chimiques usuels fournissent de bonnes prédictions, reflet de décennies d’optimisation de ces mesures. Les indices basés sur les liaisons, pris isolément, donnent des performances sensiblement inférieures, montrant qu’ils ne remplacent pas complètement les caractéristiques standard. Toutefois, lorsque les indices basés sur les liaisons sont combinés aux descripteurs physico‑chimiques, la qualité prédictive globale s’améliore : la précision moyenne sur les jeux de test augmente légèrement et un score d’erreur sans unité diminue d’environ trois pour cent. Les gains sont les plus visibles pour des grandeurs sensibles à la structure comme le nombre de liaisons rotatives et un score de « ressemblance aux produits naturels », où la connectivité détaillée compte clairement. Pour la puissance antibactérienne, tous les modèles restent modestes, ce qui suggère que des informations encore plus riches sont nécessaires pour capturer l’activité biologique complexe.
Message à retenir pour les non‑spécialistes
Cette étude montre que traiter les liaisons chimiques comme des éléments de première classe dans les descriptions moléculaires peut fournir des informations supplémentaires utiles aux modèles informatiques, surtout lorsqu’on les mélange avec des propriétés chimiques traditionnelles et globales. Les nouveaux indices sensibles aux liaisons sont bien comportés mathématiquement, faciles à calculer et aident à capturer des différences structurelles subtiles entre molécules. S’ils ne résolvent pas à eux seuls la découverte de médicaments, ils offrent une couche pratique supplémentaire de détail structural qui peut améliorer modestement mais de manière constante les prédictions dans la modélisation multi‑propriété de composés antibactériens.
Citation: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Mots-clés: chimiinformatique, descripteurs moléculaires, théorie des graphes, QSAR QSPR, découverte d’antibiotiques