Clear Sky Science · pl
Od teorii grafów do chemoinformatyki: zmodyfikowane wskaźniki oparte na wiązaniach i hipotezowy, wielozadaniowy benchmark QSAR/QSPR
Dlaczego małe połączenia molekularne mają znaczenie
Chemicy często opisują cząsteczki tak, jakby były małymi miastami: atomy to budynki, a wiązania to drogi. Przez dekady większość narzędzi matematycznych służących do przewidywania zachowania cząsteczki skupiała się na tym, co dzieje się przy „budynkach”, zamiast na „drogach” między nimi. W artykule pada proste, lecz istotne pytanie: co jeśli zwrócimy większą uwagę na same wiązania, i czy ta dodatkowa szczegółowość może pomóc komputerom lepiej przewidywać zachowanie potencjalnych antybakteryjnych leków?
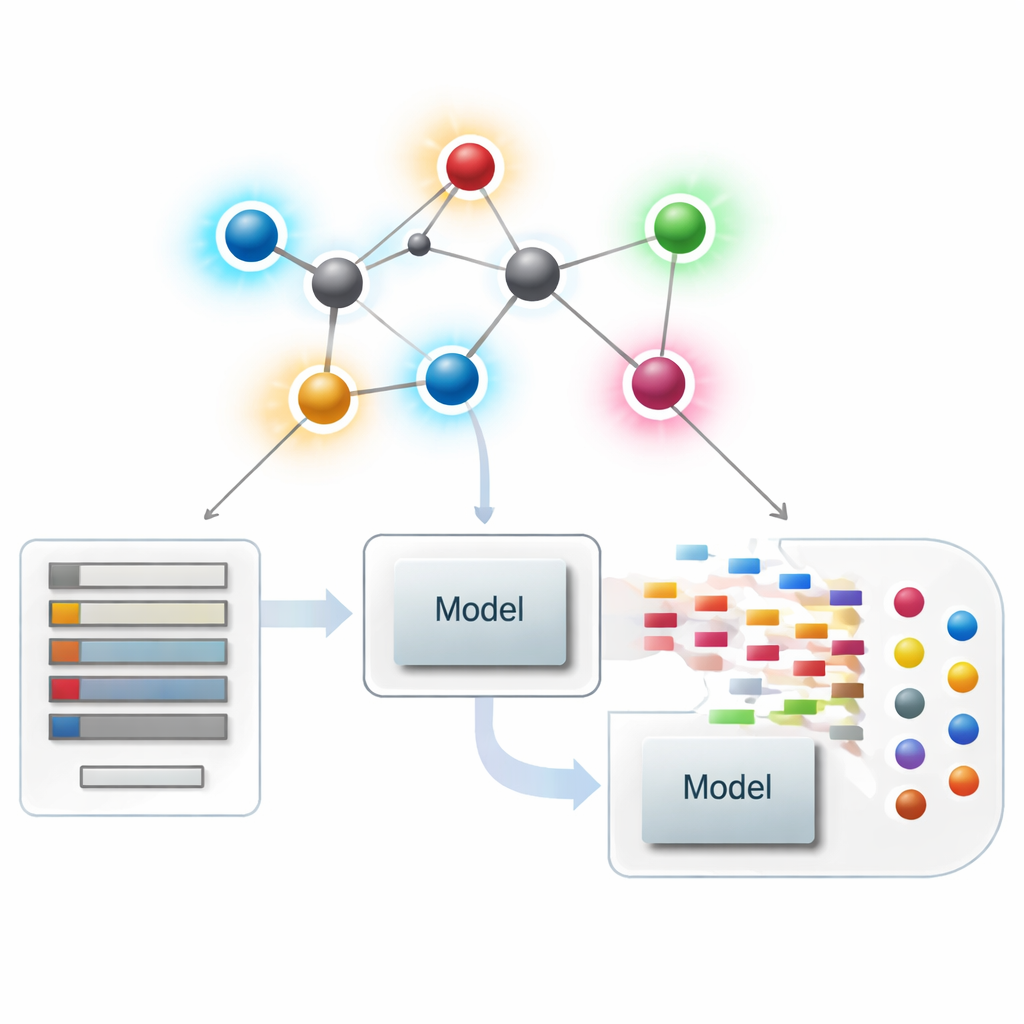
Postrzeganie cząsteczek jako sieci
W współczesnej chemoinformatyce cząsteczkę można traktować jako sieć, w której każdy atom jest węzłem, a każde wiązanie — krawędzią. Z takich sieci naukowcy wyliczają numeryczne podsumowania — zwane indeksami lub deskryptorami — które oddają aspekty kształtu, rozgałęzienia i łączności cząsteczek. Klasyczne deskryptory skupiają się głównie na tym, ile wiązań dotyka danego atomu, czyli na jego stopniu. Te deskryptory zorientowane na atomy dobrze sprawdzają się w łączeniu struktury z własnościami takimi jak temperatura wrzenia, rozpuszczalność czy „drug‑likeness”, ale mogą przeoczyć subtelne różnice między cząsteczkami, które na poziomie globalnym wyglądają podobnie, lecz zachowują się zupełnie inaczej.
Wystawiając wiązania na pierwszy plan
Autorzy wprowadzają nową rodzinę „zmodyfikowanych wskaźników opartych na wiązaniach”, które celowo przesuwają uwagę z atomów na wiązania. Dla każdego wiązania w sieci molekularnej analizują stopnie dwóch atomów, które ono łączy, i łączą je w lokalny czynnik wiązaniowy mierzący, jak zatłoczone jest otoczenie tego wiązania. Ten czynnik następnie skalibruje różne dobrze znane formuły oparte na stopniach. W efekcie każde wiązanie otrzymuje ocenę odzwierciedlającą zarówno jego końce, jak i otaczające je zagęszczenie. Wiązania w zatłoczonych obszarach cząsteczki są osłabiane, podczas gdy wiązania w spokojniejszych rejonach zyskują nieco większe znaczenie, dzięki czemu ogólny deskryptor staje się bardziej czuły na lokalne przestawienia, takie jak różne ułożenia łańcuchów bocznych.
Testowanie matematyki na zidealizowanych sieciach
Zanim zastosowano te nowe wskaźniki do rzeczywistych cząsteczek, zespół przeanalizował je na standardowych rodzinach zidealizowanych sieci dobrze znanych matematyków: ścieżkach, cyklach, grafach pełnych, gwiazdach oraz kilku bardziej złożonych strukturach‑gadżetach. Dla każdego z szesnastu zmodyfikowanych wskaźników opartych na wiązaniach wyprowadzili zwarte wzory opisujące, jak wskaźnik rośnie w miarę powiększania się lub zwiększania łączności tych sieci. Udowodnili też ostre granice łączące wartości wskaźników z podstawowymi cechami, takimi jak liczba połączeń najmniej i najbardziej połączonych węzłów. Wyniki matematyczne pokazują, że nowe deskryptory skoncentrowane na wiązaniach zachowują się w sposób kontrolowany i przewidywalny, a często sprowadzają się do prostych przeskalowań na bardzo regularnych strukturach, co ułatwia ich interpretację i porównanie ze starszymi indeksami.
Zastosowanie nowych ocen wiązań w modelowaniu leków
Mając opracowaną teorię, autorzy sprawdzili, czy te deskryptory zorientowane na wiązania rzeczywiście pomagają w praktyce. Zgromadzili skuratorowaną pulę 3 219 antybakteryjnych cząsteczek z bazy ChEMBL i rozważyli dziesięć ciągłych celów: dziewięć podstawowych wielkości fizykochemicznych (takich jak masa cząsteczkowa, polarność, pole powierzchni oraz liczba donorów i akceptorów wiązań wodorowych) oraz miarę siły antybakteryjnej. Następnie zbudowali duże „zoo modeli” regresyjnych — od prostych dopasowań liniowych po nowoczesne algorytmy drzewiaste i boosting — i porównali trzy scenariusze: użycie tylko nowych wskaźników opartych na wiązaniach, użycie tylko standardowych właściwości fizykochemicznych oraz użycie obu zestawów razem.
Co mówią wyniki o deskryptorach uwzględniających wiązania
We wszystkich dziesięciu celach standardowe deskryptory fizykochemiczne dawały silne prognozy, co odzwierciedla dekady optymalizacji tych miar. Same wskaźniki oparte na wiązaniach wypadły zauważalnie gorzej, pokazując, że nie zastępują one w pełni standardowych cech. Jednak gdy wskaźniki oparte na wiązaniach zostały połączone z deskryptorami fizykochemicznymi, ogólna jakość predykcji poprawiła się: średnia dokładność testowa wśród celów wzrosła nieznacznie, a bezwymiarowy wskaźnik błędu zmniejszył się o około trzy procent. Korzyści były najbardziej widoczne dla wielkości wrażliwych na strukturę, takich jak liczba wiązań rotowalnych i ocena „podobieństwa do produktów naturalnych”, gdzie szczegółowe połączenia mają wyraźne znaczenie. Dla mocy antybakteryjnej wszystkie modele pozostały umiarkowane, co sugeruje, że do uchwycenia złożonej aktywności biologicznej potrzebne są jeszcze bogatsze informacje.
Wniosek dla osób niebędących specjalistami
Badanie pokazuje, że traktowanie wiązań chemicznych jako pełnoprawnych elementów opisu molekularnego może dostarczyć dodatkowych, użytecznych informacji dla modeli komputerowych, szczególnie gdy są one połączone z tradycyjnymi, „masowymi” właściwościami chemicznymi. Nowe wskaźniki uwzględniające wiązania cechują się dobrą postawą matematyczną, są łatwe do obliczenia i pomagają uchwycić subtelne różnice strukturalne między cząsteczkami. Chociaż same w sobie nie rozwiązują procesu odkrywania leków, oferują praktyczną, nową warstwę szczegółu strukturalnego, która może skromnie, lecz konsekwentnie poprawiać prognozy w modelowaniu wielowłaściwościowym związków antybakteryjnych.
Cytowanie: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Słowa kluczowe: chemoinformatyka, deskryptory molekularne, teoria grafów, QSAR QSPR, poszukiwanie antybakteryjnych leków