Clear Sky Science · de
Von der Graphentheorie zur Chemoinformatik: modifizierte bindungsbasierte Indizes und ein hypothesengesteuerter Multi‑Task‑QSAR/QSPR‑Benchmark
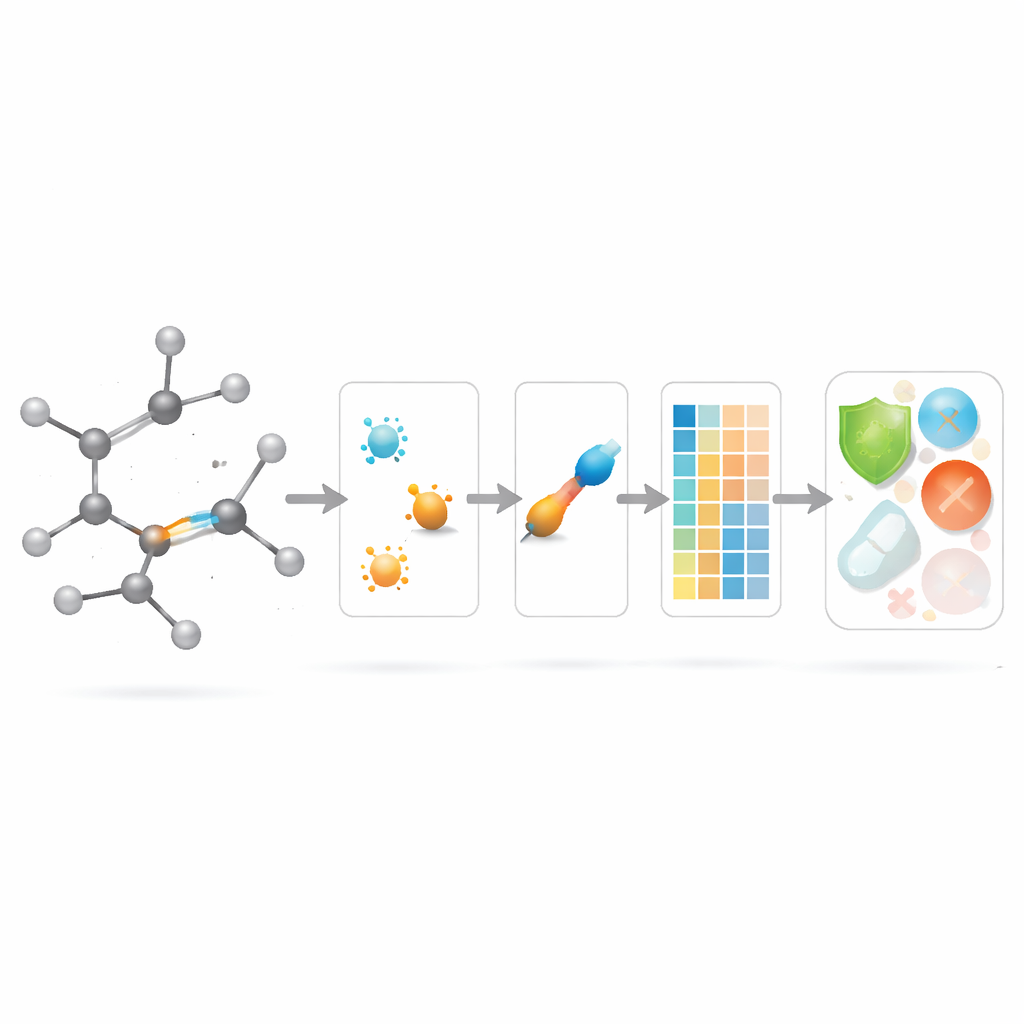
Warum winzige molekulare Verbindungen wichtig sind
Chemiker beschreiben Moleküle oft, als wären sie winzige Städte: Atome sind die Gebäude und Bindungen die Straßen. Jahrzehntelang konzentrierten sich die meisten mathematischen Werkzeuge zur Vorhersage des Verhaltens eines Moleküls darauf, was an den „Gebäuden“ passiert, statt auf die „Straßen“ dazwischen. Dieses Papier stellt eine einfache, aber einflussreiche Frage: Was, wenn wir den Bindungen selbst mehr Aufmerksamkeit schenken — und kann diese zusätzliche Detailgenauigkeit Computern helfen, besser vorherzusagen, wie potenzielle antibakterielle Wirkstoffe wirken?
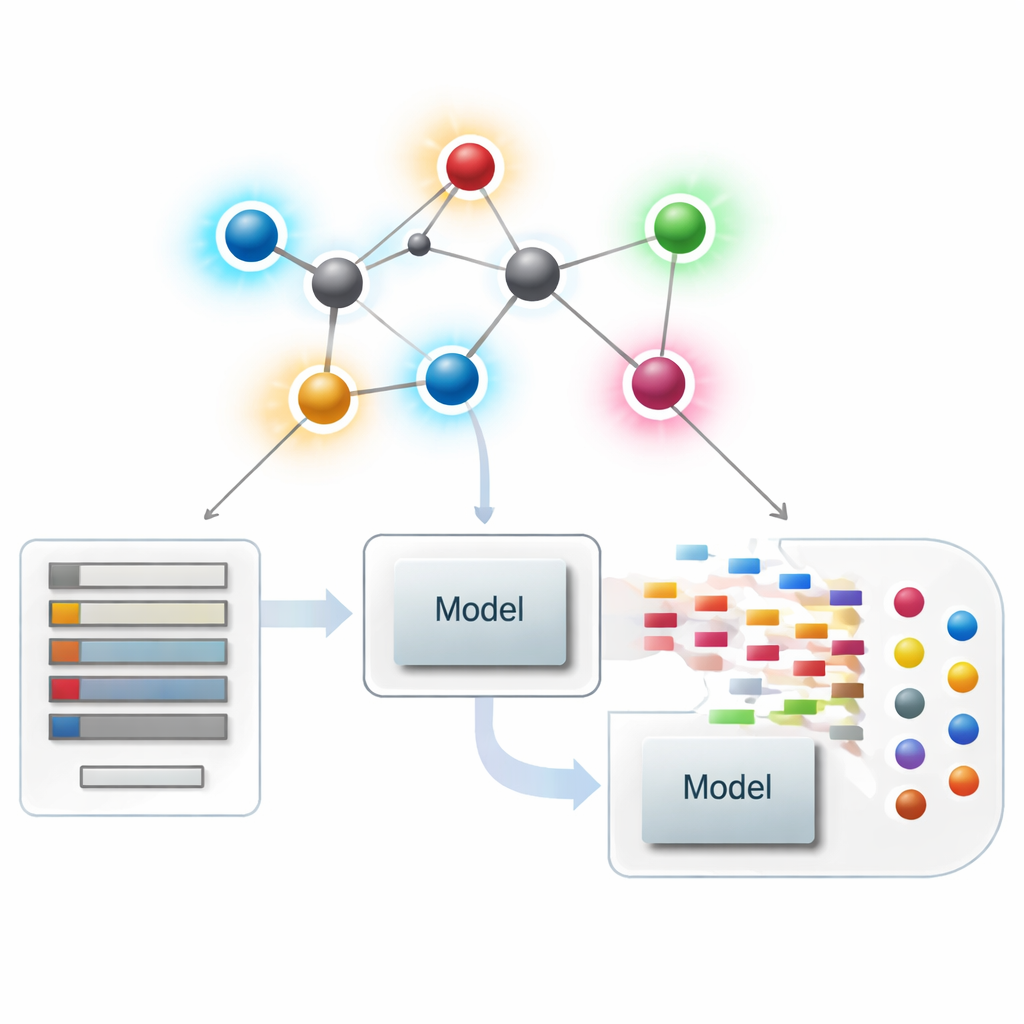
Moleküle als Netzwerke betrachten
In der modernen Chemoinformatik kann ein Molekül als Netzwerk behandelt werden, bei dem jedes Atom ein Knoten und jede chemische Bindung eine Kante ist. Aus diesen Netzwerken berechnen Forscher numerische Zusammenfassungen — so genannte Indizes oder Deskriptoren —, die Aspekte von Molekülgestalt, Verzweigung und Konnektivität erfassen. Klassische Deskriptoren konzentrieren sich überwiegend darauf, wie viele Bindungen an jedes Atom angrenzen, eine Größe, die als Grad bezeichnet wird. Diese atomzentrierten Zusammenfassungen waren sehr erfolgreich, um Struktur mit Eigenschaften wie Siedepunkt, Löslichkeit oder Drug‑Likeness in Verbindung zu bringen, können aber feine Unterschiede zwischen Molekülen übersehen, die global ähnlich aussehen, sich jedoch sehr unterschiedlich verhalten.
Bindungen ins Rampenlicht rücken
Die Autoren führen eine neue Familie von „modifizierten bindungsbasierten Indizes“ ein, die bewusst die Aufmerksamkeit von Atomen auf Bindungen verlagern. Für jede Bindung in einem molekularen Netzwerk betrachten sie die Grade der beiden daran beteiligten Atome und kombinieren diese zu einem lokalen Bindungsfaktor, der misst, wie dicht bevölkert die Nachbarschaft der Bindung ist. Dieser Faktor skaliert dann eine Vielzahl vertrauter, gradbasierter Formeln. Effektiv erhält jede Bindung eine Punktzahl, die sowohl ihre Endpunkte als auch die umliegende Überfüllung widerspiegelt. Bindungen in dicht besetzten Regionen eines Moleküls werden abgeschwächt gewichtet, während Bindungen in ruhigeren Bereichen etwas stärker zählen, wodurch der Gesamt‑Deskriptor empfindlicher auf lokale Umordnungen wie unterschiedliche Seitenkettenanordnungen reagiert.
Mathematik an idealisierten Netzwerken prüfen
Bevor diese neuen Indizes an realen Molekülen angewandt werden, analysiert das Team sie auf standardisierten Familien idealisierter Netzwerke, die Mathematiker gut kennen: Pfade, Zyklen, vollständige Graphen, Sterne und mehrere elaboriertere „Gadget“-Strukturen. Für sechzehn modifizierte bindungsbasierte Indizes leiten sie kompakte Formeln her, die angeben, wie der Index wächst, wenn diese Netzwerke größer oder stärker verbunden werden. Sie beweisen auch scharfe Schranken, die Indexwerte mit grundlegenden Merkmalen verknüpfen, etwa wie viele Verbindungen die am wenigsten bzw. am stärksten verbundenen Knoten haben. Diese mathematischen Ergebnisse zeigen, dass die neuen bindungsfokussierten Deskriptoren sich kontrolliert und vorhersehbar verhalten und sich auf sehr regulären Strukturen oft zu einfachen Reskalierungen reduzieren, was ihre Interpretation und den Vergleich mit älteren Indizes erleichtert.
Neue Bindungsbewertungen in der Wirkstoffmodellierung einsetzen
Mit der Theorie im Hintergrund fragen die Autoren, ob diese bindungszentrierten Deskriptoren in der Praxis tatsächlich nützen. Sie stellen einen kuratierten Satz von 3.219 antibakteriellen Molekülen aus der ChEMBL‑Datenbank zusammen und betrachten zehn kontinuierliche Ziele: neun grundlegende physikochemische Größen (wie Molekulargewicht, Polarität, Oberfläche und Zählungen von Wasserstoffbrücken‑Donoren und ‑Akzeptoren) sowie eine Messgröße für antibakterielle Wirksamkeit. Anschließend bauen sie einen großen „Model Zoo“ von Regressionsmethoden auf, von einfachen linearen Anpassungen bis zu modernen baumbasierten und Boosting‑Algorithmen, und vergleichen drei Szenarien: nur die neuen bindungsbasierten Indizes verwenden, nur standardmäßige physikochemische Eigenschaften verwenden und beides zusammen.
Was die Ergebnisse über bindungsbewusste Deskriptoren aussagen
Über alle zehn Ziele hinweg liefern die üblichen physikochemischen Deskriptoren starke Vorhersagen, was Jahrzehnte der Optimierung solcher Maße widerspiegelt. Die bindungsbasierten Indizes allein schneiden deutlich schlechter ab, was zeigt, dass sie kein vollständiger Ersatz für die Standardmerkmale sind. Werden die bindungsbasierten Indizes jedoch mit physikochemischen Deskriptoren kombiniert, verbessert sich die Gesamtvorhersagequalität: Die durchschnittliche Testgenauigkeit über die Ziele steigt leicht und ein einheitenfreier Fehlermaßstab verringert sich um etwa drei Prozent. Die Verbesserungen sind am sichtbarsten für struktur‑sensitive Größen wie die Anzahl der rotierbaren Bindungen und einen „Natural‑Product‑Likeness“-Score, bei denen detaillierte Konnektivität eindeutig eine Rolle spielt. Für die antibakterielle Potenz bleiben alle Modelle mäßig, was darauf hindeutet, dass noch reichhaltigere Informationen nötig sind, um komplexe biologische Aktivität zu erfassen.
Kernaussage für Nicht‑Spezialisten
Die Studie zeigt, dass die Behandlung chemischer Bindungen als gleichberechtigte Elemente molekularer Beschreibungen zusätzliche, nützliche Informationen für Computermodelle liefern kann, insbesondere in Kombination mit traditionellen, makroskopischen chemischen Eigenschaften. Die neuen bindungsbewussten Indizes sind mathematisch gutartig, leicht zu berechnen und helfen, subtile strukturelle Unterschiede zwischen Molekülen einzufangen. Zwar lösen sie die Wirkstoffforschung nicht allein, bieten aber eine praktische neue Ebene struktureller Details, die Vorhersagen in Multi‑Eigenschafts‑Modellen für antibakterielle Verbindungen moderat, aber konsistent verbessern kann.
Zitation: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Schlüsselwörter: Chemoinformatik, molekulare Deskriptoren, Graphentheorie, QSAR QSPR, Entdeckung antibakterieller Wirkstoffe