Clear Sky Science · it
Dalla teoria dei grafi alla chemoinformatica: indici modificati basati sui legami e un benchmark QSAR/QSPR multitask guidato da ipotesi
Perché contano le piccole connessioni molecolari
I chimici spesso descrivono le molecole come se fossero piccole città: gli atomi sono gli edifici e i legami sono le strade. Per decenni, la maggior parte degli strumenti matematici per prevedere il comportamento di una molecola si è concentrata sul contare ciò che avviene negli “edifici” piuttosto che sulle “strade” che li collegano. Questo articolo pone una domanda semplice ma potente: se prestassimo maggiore attenzione ai legami stessi, quel dettaglio aggiuntivo potrebbe aiutare i computer a prevedere meglio il comportamento di potenziali farmaci antibatterici?
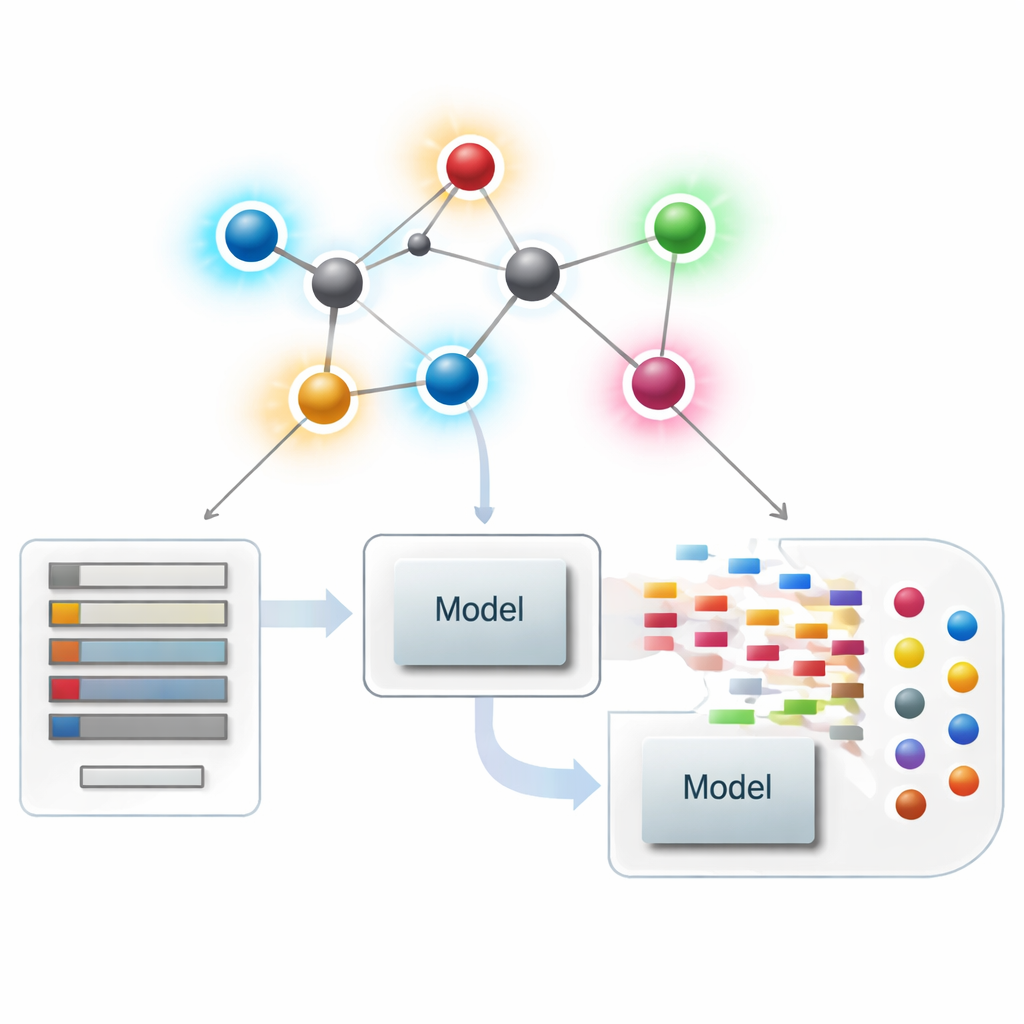
Guardare le molecole come reti
Nella chemoinformatica moderna, una molecola può essere trattata come una rete, dove ogni atomo è un punto e ogni legame chimico è una linea. Da queste reti, gli scienziati calcolano sintesi numeriche—chiamate indici o descrittori—che catturano aspetti della forma molecolare, della ramificazione e della connettività. I descrittori classici si concentrano soprattutto su quante connessioni toccano ogni atomo, una quantità chiamata grado. Questi riassunti centrati sugli atomi hanno avuto grande successo nel correlare struttura e proprietà come punto di ebollizione, solubilità o affinità da farmaco, ma possono non cogliere differenze sottili tra molecole che appaiono simili a livello globale ma agiscono in modo molto diverso.
Mettere i legami sotto i riflettori
Gli autori introducono una nuova famiglia di “indici modificati basati sui legami” che spostano deliberatamente l’attenzione dagli atomi ai legami. Per ogni legame in una rete molecolare, considerano i gradi dei due atomi che collega e li combinano in un fattore locale del legame che misura quanto sia affollato il vicinato del legame. Questo fattore poi scala una varietà di formule familiari basate sui gradi. In pratica, a ogni legame viene assegnato un punteggio che riflette sia i suoi endpoint sia la congestione circostante. I legami nelle regioni più affollate della molecola sono pesati verso il basso, mentre i legami in regioni più tranquille contano un po’ di più, rendendo il descrittore complessivo più sensibile a riorganizzazioni locali come diverse disposizioni delle catene laterali.
Testare la matematica su reti idealizzate
Prima di utilizzare questi nuovi indici su molecole reali, il team li analizza su famiglie standard di reti idealizzate note ai matematici: cammini, cicli, grafi completi, stelle e diverse strutture «gadget» più elaborate. Per ciascuno dei sedici indici modificati basati sui legami, ricavano formule compatte che indicano come l’indice cresce quando queste reti diventano più grandi o più connesse. Dimostrano anche limiti netti che collegano i valori degli indici a caratteristiche di base come il numero di connessioni dei nodi meno e più connessi. Questi risultati matematici mostrano che i nuovi descrittori focalizzati sui legami si comportano in modo controllato e prevedibile e spesso si riducono a semplici riscalature su strutture molto regolari, il che aiuta a interpretarli e a confrontarli con indici più vecchi.
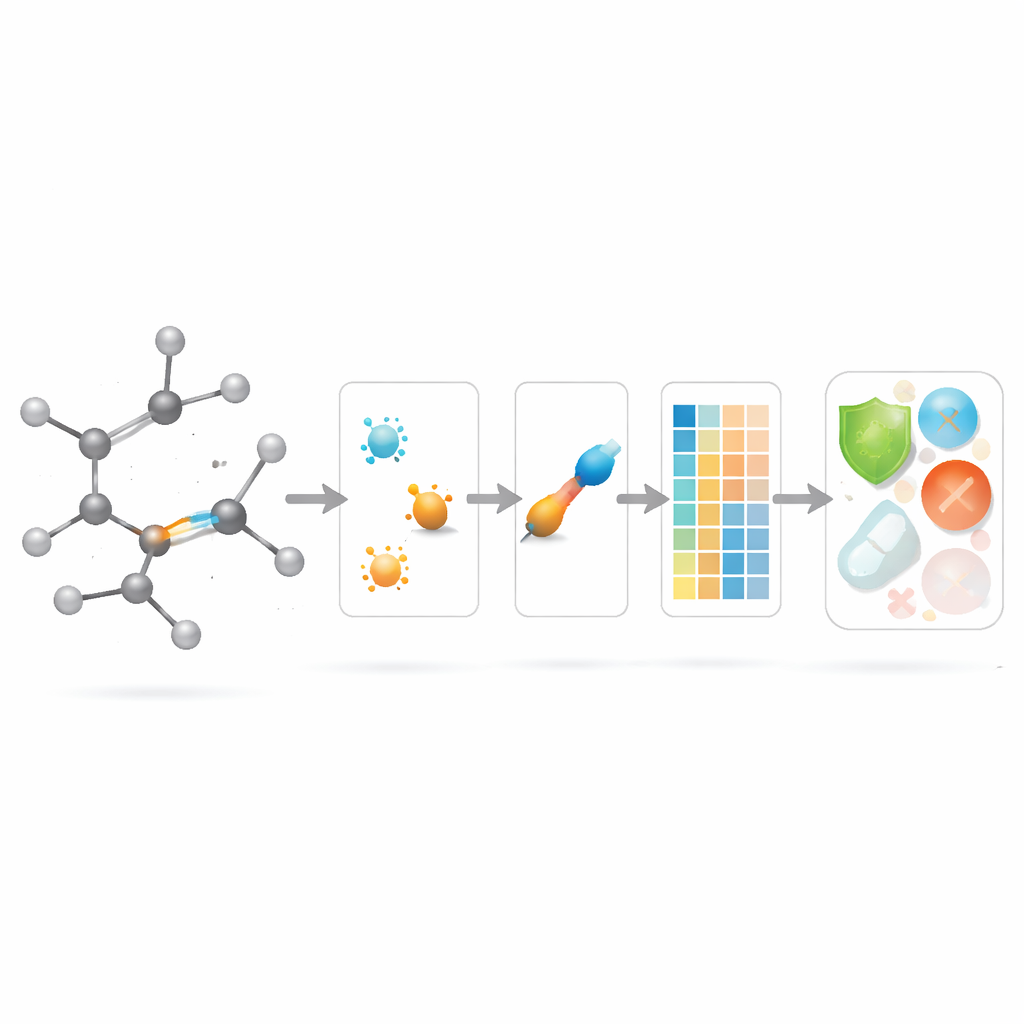
Applicare i nuovi punteggi dei legami nella modellazione farmaceutica
Con la teoria definita, gli autori si chiedono se questi descrittori centrati sui legami siano effettivamente utili in pratica. Assemblano un set curato di 3.219 molecole antibatteriche dal database ChEMBL e considerano dieci target continui: nove quantità fisico-chimiche di base (come peso molecolare, polarità, area superficiale e conteggi di donatori e accettori di legami idrogeno) oltre a una misura di potenza antibatterica. Costruiscono poi un ampio “zoo di modelli” di metodi di regressione, da semplici aggiustamenti lineari ad algoritmi moderni basati su alberi e boosting, e confrontano tre scenari: usare solo i nuovi indici basati sui legami, usare solo le proprietà fisico-chimiche standard e usare entrambi insieme.
Cosa dicono i risultati sui descrittori sensibili ai legami
Su tutti e dieci i target, i descrittori fisico-chimici usuali forniscono previsioni solide, riflettendo decenni di ottimizzazione di tali misure. Gli indici basati sui legami da soli si comportano visibilmente peggio, mostrando che non sono un sostituto completo delle caratteristiche standard. Tuttavia, quando gli indici basati sui legami sono combinati con i descrittori fisico-chimici, la qualità complessiva delle predizioni migliora: l’accuratezza media sul test aumenta leggermente e un punteggio di errore senza unità diminuisce di circa il tre percento. I guadagni sono più visibili per quantità sensibili alla struttura come il numero di legami rotabili e un punteggio di «natural product‑likeness», dove la connettività dettagliata è chiaramente importante. Per la potenza antibatterica, tutti i modelli rimangono modesti, suggerendo che sono necessarie informazioni ancora più ricche per catturare l’attività biologica complessa.
Messaggio principale per i non specialisti
Questo studio mostra che trattare i legami chimici come cittadini di prima classe nelle descrizioni molecolari può fornire informazioni extra e utili per i modelli computazionali, soprattutto se miscelate con le proprietà chimiche tradizionali e globali. I nuovi indici sensibili ai legami sono matematicamente ben comportati, facili da calcolare e aiutano a catturare differenze strutturali sottili tra molecole. Pur non risolvendo la scoperta di farmaci da soli, offrono un nuovo strato pratico di dettaglio strutturale che può migliorare in modo modesto ma consistente le predizioni nella modellazione multitarget di composti antibatterici.
Citazione: Altairi, A., Alhaj, Z., Alsharafi, M. et al. From graph theory to chemoinformatics: modified bond-based indices and a hypothesis-driven multi-task QSAR/QSPR benchmark. Sci Rep 16, 10104 (2026). https://doi.org/10.1038/s41598-026-40969-7
Parole chiave: chemoinformatica, descrittori molecolari, teoria dei grafi, QSAR QSPR, scoperta di farmaci antibatterici