Clear Sky Science · zh
内皮细胞中IL-32γ上调促进巨噬细胞募集和动脉粥样硬化炎性进展
这项研究为何对心脏健康重要
动脉粥样硬化——动脉堵塞和硬化——是大多数心肌梗死和中风的根本原因。我们知道胆固醇,尤其是其受损形式,以及慢性炎症都会推动这一疾病的发展,但这两种力量在血管内如何相互作用仍在研究之中。本研究聚焦于一种鲜为人知的炎症介质IL‑32γ,展示了它如何将受压的血管内皮细胞转变为对有害免疫细胞具有巨大吸引力和放大作用的“磁铁”,可能为在源头上抑制动脉炎症打开一条新路径。
细看堵塞的动脉
动脉内壁由一层薄薄的内皮细胞构成,通常在血液与血管壁之间形成光滑的保护屏障。当“坏”胆固醇水平升高时,部分颗粒会滑入这层内皮下并被氧化——成为化学损伤的氧化低密度脂蛋白(Ox‑LDL)。这种氧化胆固醇刺激内皮细胞,使其变得渗漏且易粘附,并吸引叫做单核细胞的免疫细胞,后者转化为吞噬脂质的巨噬细胞。随着时间推移,这些脂质吞噬细胞连同瘢痕组织积聚成斑块,斑块可能突然破裂,阻断流向心脏或大脑的血液。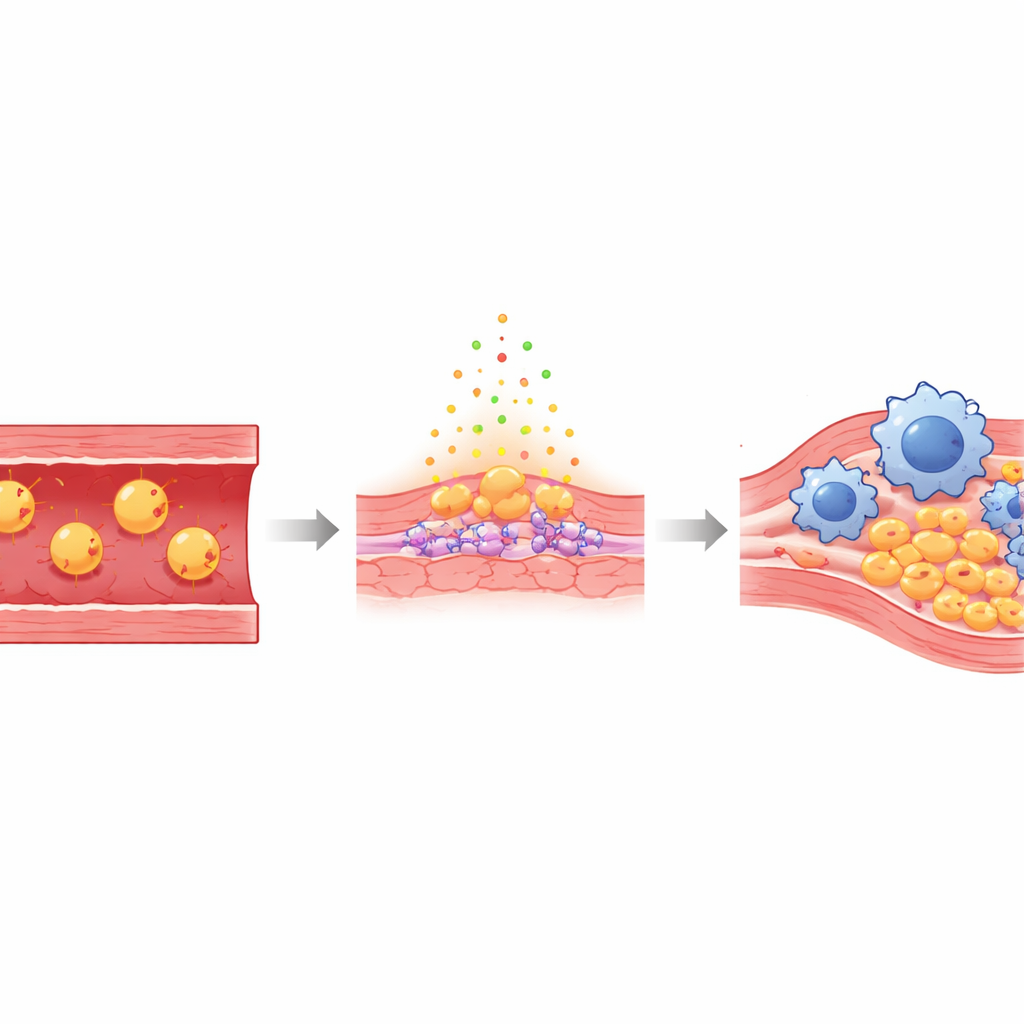
处于中心的炎症信使
研究人员关注IL‑32,这是一种存在多种亚型的炎症蛋白,其中IL‑32γ被认为活性最强。研究者首先测量了动脉粥样硬化患者和健康志愿者血液中不同IL‑32变体的水平。只有IL‑32γ在患者中在基因和蛋白水平上明显升高,提示它可能与动脉疾病特别相关。随后他们在体外用Ox‑LDL处理人类内皮细胞。在这种压力下,细胞显著增加IL‑32γ的产生和释放,而其他IL‑32形式几乎没有变化。与此同时,细胞的增殖、迁移和形成类血管结构的能力减弱,并上调了帮助捕获经过免疫细胞的分子——这些都是内皮功能障碍的典型特征。
血管细胞如何将巨噬细胞推向有害状态
为了观察这对免疫行为的影响,团队在能通过可溶性信号相互通讯的Transwell系统中将经Ox‑LDL处理的内皮细胞与巨噬细胞共培养。暴露于受压内皮细胞的巨噬细胞向强烈促炎的“M1”表型转变:它们表现出与攻击模式相关的表面标志,产生更多如TNF‑α和IL‑1β的炎性物质,并更容易向内皮层迁移。当研究人员阻断内皮细胞内的关键开关NF‑κB通路时,既抑制了IL‑32γ的释放,也减弱了这种M1偏向。直接中和IL‑32γ本身产生了类似效果,降低了炎性标志并抑制了巨噬细胞的迁移,指向IL‑32γ作为关键的中介者。
聚焦信号级联
科学家接着询问IL‑32γ是否能直接作用于巨噬细胞以及通过何种途径。向巨噬细胞添加纯化的IL‑32γ(不加入任何内皮细胞)就足以将其推入M1状态并增加迁移。该反应依赖于细胞内的另一路分子传导——p38 MAPK通路:当加入p38抑制药时,IL‑32γ不再促进炎性基因表达、细胞因子释放或迁移。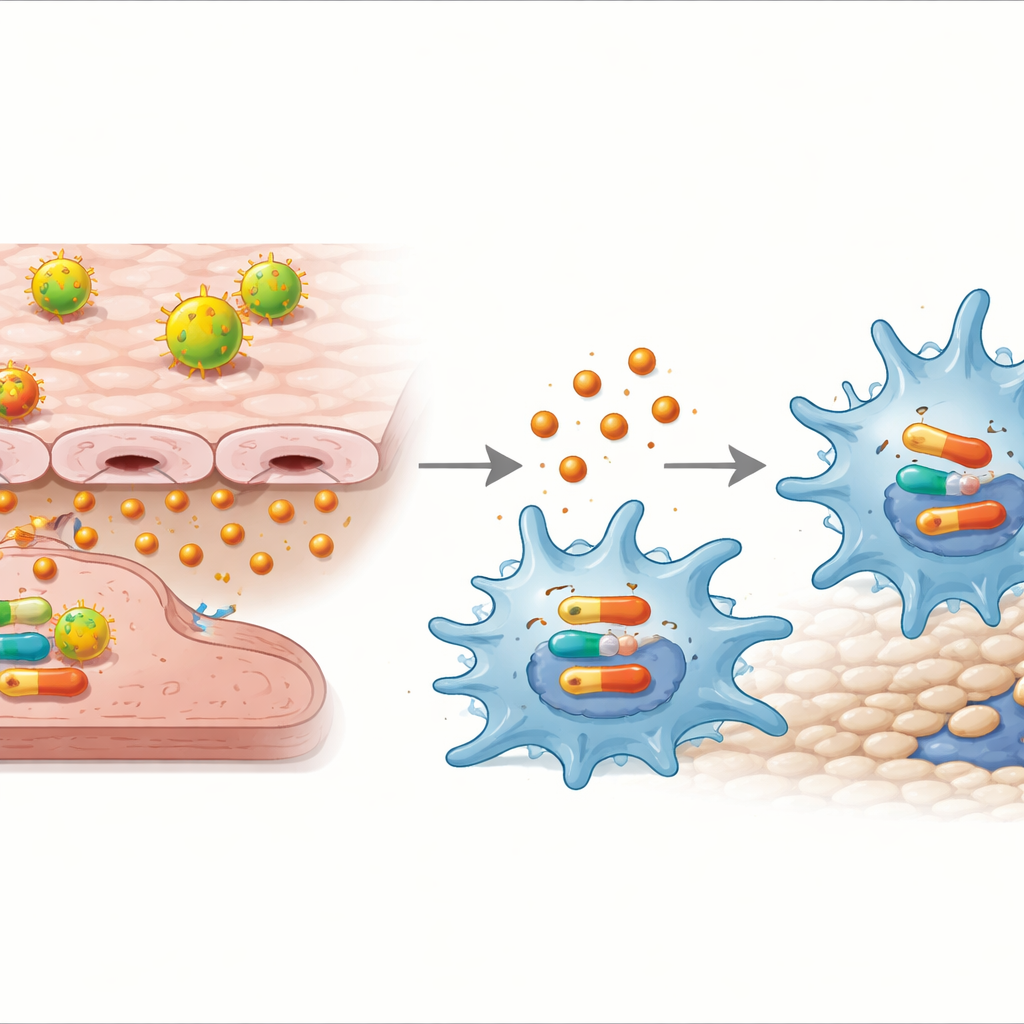
活体动脉中发生了什么
最后,团队在遗传上易患动脉粥样硬化的小鼠中并给予西式高脂饮食来检验该通路。注射IL‑32γ的小鼠形成了更大的动脉斑块,斑块内巨噬细胞堆积更多,血液中的炎性分子水平也更高——尽管与对照组相比胆固醇水平相似。当在这些动物中阻断p38通路时,斑块大小、巨噬细胞积聚和炎症水平均回落至接近基线。这表明IL‑32γ主要通过增强炎性信号而非改变血脂来加速活体动脉内的斑块生长。
对未来治疗的意义
综合来看,这项工作描绘了一条事件链:氧化胆固醇刺激血管内皮,内皮细胞通过释放IL‑32γ作出反应,而IL‑32γ又通过p38信号将周围巨噬细胞驱动到具有侵袭性、促进斑块的状态。将IL‑32γ确定为受损内皮细胞与过度活跃免疫细胞之间的关键纽带,提示了一种新的治疗思路:除了单纯降低胆固醇外,未来药物也可针对IL‑32γ或其下游的p38通路,以冷却动脉壁内的炎症,减缓甚至可能逆转动脉粥样硬化的进展。
引用: Yang, Z., Liu, M., Shi, Y. et al. Upregulation of IL-32γ in endothelial cells promotes macrophage recruitment and inflammatory progression in atherosclerosis. Sci Rep 16, 11262 (2026). https://doi.org/10.1038/s41598-026-40151-z
关键词: 动脉粥样硬化, 内皮细胞, 巨噬细胞极化, 炎症, IL-32 γ