Clear Sky Science · en
Upregulation of IL-32γ in endothelial cells promotes macrophage recruitment and inflammatory progression in atherosclerosis
Why this research matters for heart health
Atherosclerosis—clogging and hardening of the arteries—is the root cause of most heart attacks and strokes. We know that cholesterol, especially in its damaged form, and chronic inflammation both drive this disease, but how these two forces talk to each other inside blood vessels is still being worked out. This study zeroes in on a little-known inflammatory messenger called IL‑32γ and shows how it helps turn stressed vessel-lining cells into powerful magnets and amplifiers for harmful immune cells, potentially opening a new way to calm arterial inflammation at its source.
A closer look at clogged arteries
Arteries are lined by a thin sheet of endothelial cells that normally form a smooth, protective barrier between blood and the vessel wall. When levels of “bad” cholesterol are high, some of these particles slip underneath this lining and become oxidized—chemically damaged versions known as Ox‑LDL. This oxidized cholesterol irritates the endothelial cells, making them leaky and sticky, and it calls in immune cells called monocytes, which turn into macrophages that gobble up fat. Over time, these fat-engorged cells, along with scar tissue, build up into plaques that can suddenly rupture, blocking blood flow to the heart or brain. 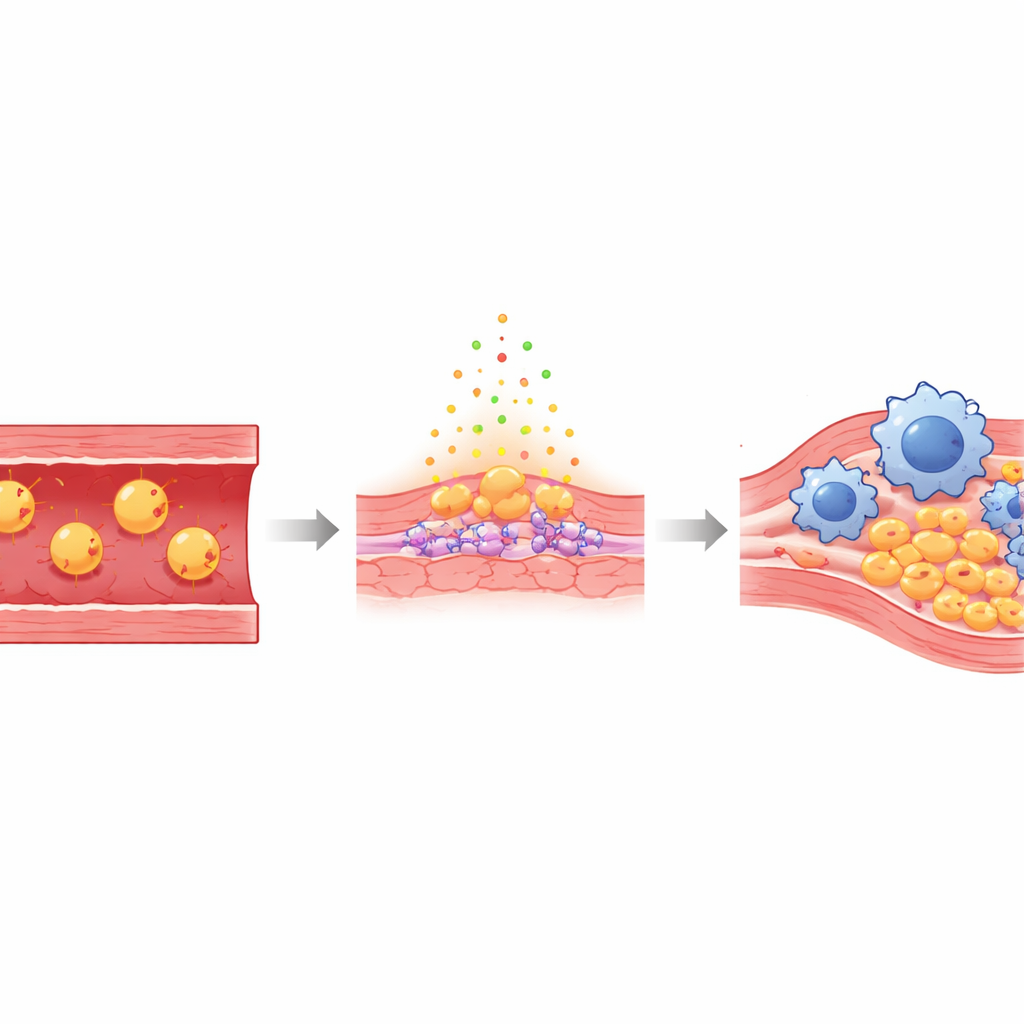
The inflammatory messenger in the middle
The researchers focused on IL‑32, an inflammatory protein that exists in several forms, with IL‑32γ considered the most active. They first measured different IL‑32 variants in blood from people with atherosclerosis and healthy volunteers. Only IL‑32γ was clearly higher in patients, at both the gene and protein levels, suggesting it might be specially linked to artery disease. They then exposed human endothelial cells to Ox‑LDL in the lab. Under this stress, the cells sharply increased production and release of IL‑32γ, while other IL‑32 forms barely changed. At the same time, the cells became less able to grow, migrate, and form new vessel-like structures, and they ramped up molecules that help grab passing immune cells—classic signs of endothelial dysfunction.
How vessel cells push macrophages toward a harmful state
To see what this meant for immune behavior, the team co-cultured Ox‑LDL‑treated endothelial cells with macrophages in a Transwell system that lets the cells communicate through soluble signals. Macrophages exposed to stressed endothelial cells shifted toward a strongly pro‑inflammatory “M1” pattern: they displayed surface markers associated with attack mode, produced more inflammatory substances like TNF‑α and IL‑1β, and migrated more readily toward the endothelial layer. When the researchers blocked a key switch inside endothelial cells, the NF‑κB pathway, they dampened both IL‑32γ release and this M1 skewing. Neutralizing IL‑32γ itself had a similar effect, reducing inflammatory markers and curbing macrophage movement, pointing to IL‑32γ as a central go‑between.
Zooming in on the signaling chain
The scientists then asked whether IL‑32γ could act directly on macrophages and by what route. Adding purified IL‑32γ to macrophages, without any endothelial cells present, was enough to push them into the M1 state and increase their movement. This response depended on another molecular relay inside the cells, the p38 MAPK pathway: when a p38‑blocking drug was added, IL‑32γ no longer boosted inflammatory genes, cytokine release, or migration. 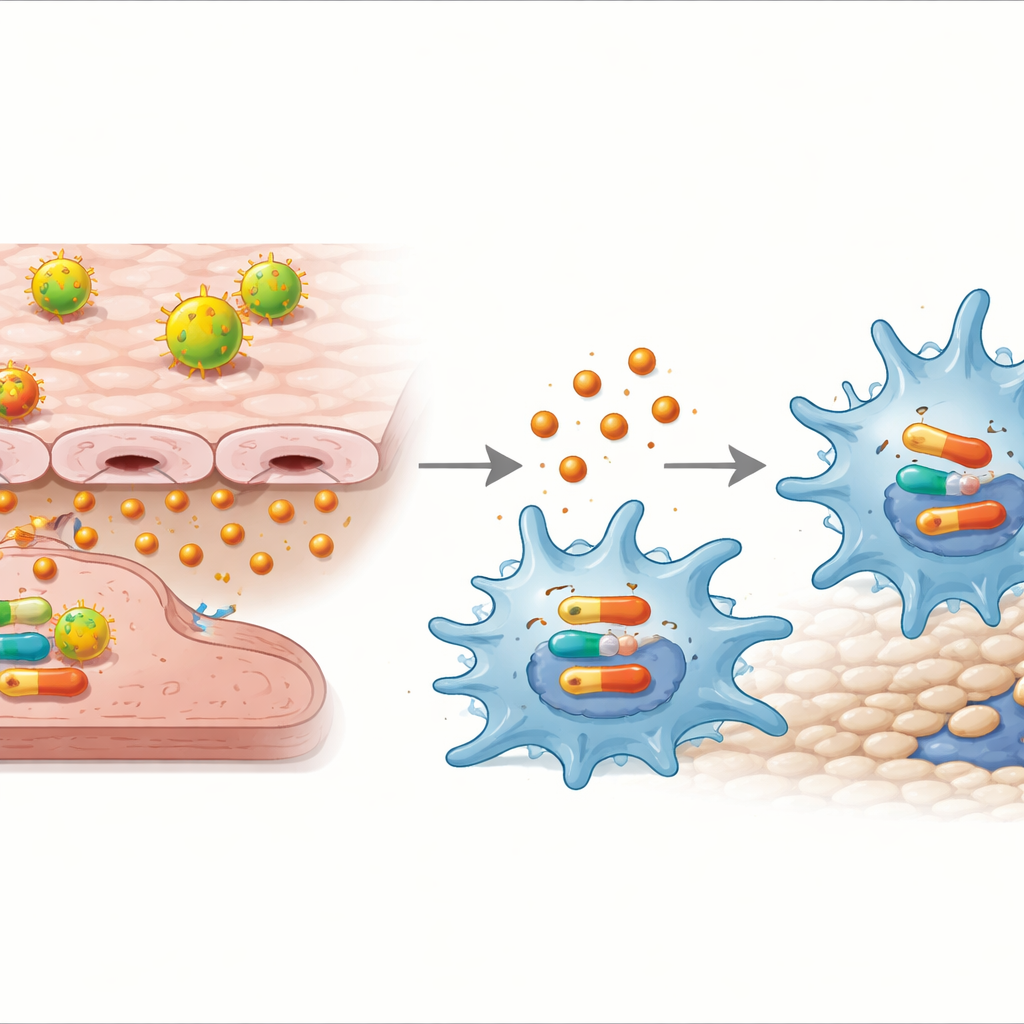
What happens in living arteries
Finally, the team tested the pathway in mice genetically prone to atherosclerosis and fed a Western‑style high‑fat diet. Mice injected with IL‑32γ developed larger arterial plaques, showed more macrophage build‑up inside those plaques, and had higher blood levels of inflammatory molecules—despite similar cholesterol levels compared to controls. When the p38 pathway was blocked in these animals, plaque size, macrophage accumulation, and inflammation dropped back toward baseline. This means IL‑32γ can speed up plaque growth in living arteries mainly by turning up inflammatory signaling, not by altering blood lipids.
What it all means for future treatments
Taken together, this work outlines a chain of events: oxidized cholesterol irritates the vessel lining, those cells respond by releasing IL‑32γ, and IL‑32γ in turn drives nearby macrophages into an aggressive, plaque‑promoting mode through p38 signaling. By identifying IL‑32γ as a key link between damaged endothelial cells and overactive immune cells, the study suggests a new therapeutic angle: instead of only lowering cholesterol, future drugs might also target IL‑32γ or its p38 pathway to cool down inflammation inside artery walls and slow, or possibly reverse, the progression of atherosclerosis.
Citation: Yang, Z., Liu, M., Shi, Y. et al. Upregulation of IL-32γ in endothelial cells promotes macrophage recruitment and inflammatory progression in atherosclerosis. Sci Rep 16, 11262 (2026). https://doi.org/10.1038/s41598-026-40151-z
Keywords: atherosclerosis, endothelial cells, macrophage polarization, inflammation, IL-32 gamma