Clear Sky Science · de
Hochregulierung von IL-32γ in Endothelzellen fördert Makrophagenrekrutierung und Entzündungsprogression bei Atherosklerose
Warum diese Forschung für die Herzgesundheit wichtig ist
Atherosklerose – Verstopfung und Verhärtung der Arterien – ist die Hauptursache der meisten Herzinfarkte und Schlaganfälle. Wir wissen, dass Cholesterin, insbesondere in seiner geschädigten Form, und chronische Entzündung die Krankheit vorantreiben, doch wie diese beiden Kräfte innerhalb der Blutgefäße miteinander kommunizieren, ist noch nicht vollständig geklärt. Diese Studie richtet den Blick auf einen wenig bekannten entzündlichen Botenstoff, IL‑32γ, und zeigt, wie er gestresste, gefäßauskleidende Zellen zu starken Anziehungs- und Verstärkerquellen für schädliche Immunzellen macht – was einen neuen Ansatz eröffnen könnte, arterielle Entzündungen an ihrer Quelle zu dämpfen.
Ein genauerer Blick auf verstopfte Arterien
Arterien sind von einer dünnen Schicht Endothelzellen ausgekleidet, die normalerweise eine glatte, schützende Barriere zwischen Blut und Gefäßwand bilden. Wenn die Konzentration des „schlechten“ Cholesterins hoch ist, gelangen einige dieser Partikel unter diese Schicht und oxidieren – chemisch geschädigte Formen, bekannt als Ox‑LDL. Dieses oxidierte Cholesterin reizt die Endothelzellen, macht sie durchlässig und klebrig und lockt Immunzellen an, sogenannte Monozyten, die sich zu Makrophagen entwickeln und Fett aufnehmen. Mit der Zeit sammeln sich diese fettreichen Zellen zusammen mit Narbengewebe zu Plaques an, die plötzlich aufreißen und den Blutfluss zum Herzen oder Gehirn blockieren können. 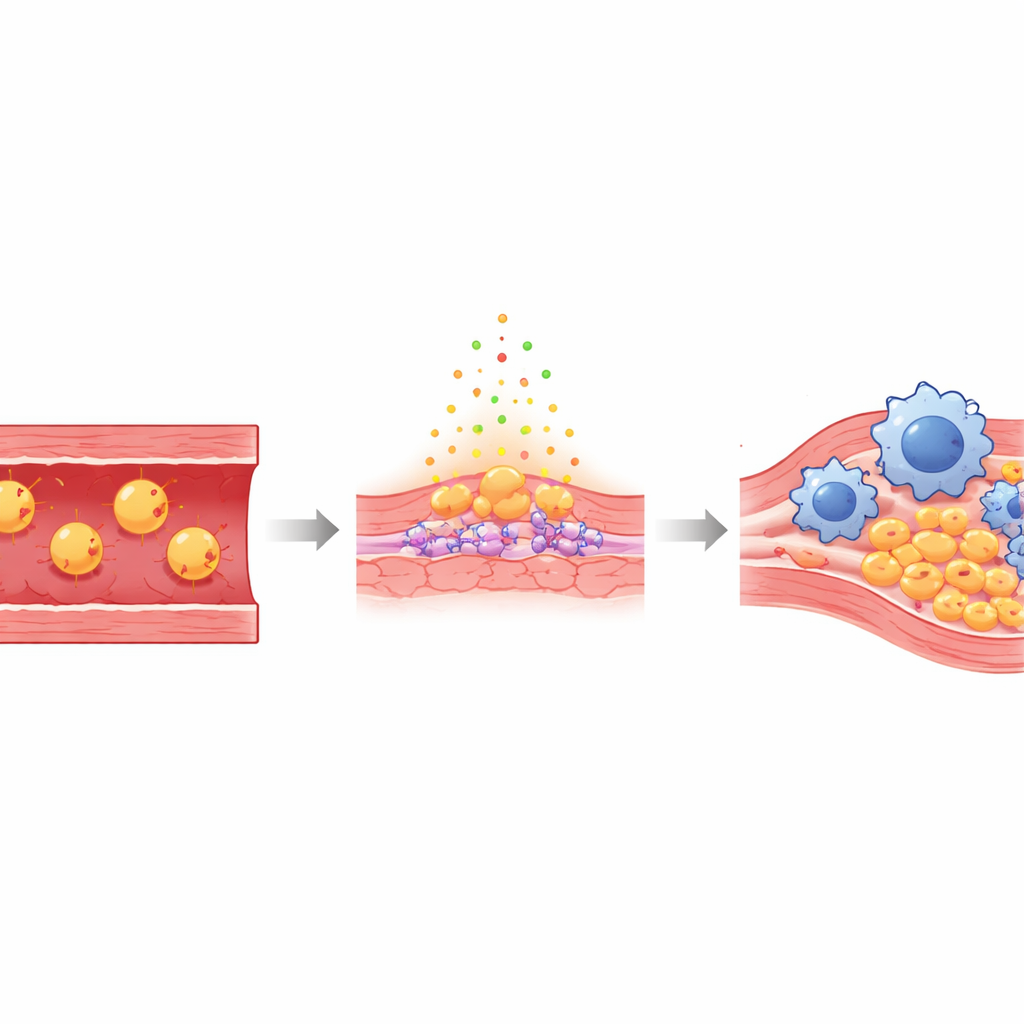
Der entzündliche Botenstoff in der Mitte
Die Forschenden konzentrierten sich auf IL‑32, ein entzündliches Protein, das in mehreren Varianten vorkommt, wobei IL‑32γ als die aktivste Form gilt. Zunächst bestimmten sie verschiedene IL‑32-Varianten im Blut von Menschen mit Atherosklerose und von gesunden Freiwilligen. Nur IL‑32γ war bei Patienten deutlich erhöht, sowohl auf Gen- als auch auf Proteinlevel, was auf eine besondere Verbindung zur Gefäßerkrankung hindeutet. Anschließend setzten sie humane Endothelzellen im Labor Ox‑LDL aus. Unter diesem Stress steigerten die Zellen die Produktion und Freisetzung von IL‑32γ deutlich, während andere IL‑32-Formen kaum verändert waren. Gleichzeitig ließen die Zellen ihre Fähigkeit zu wachsen, zu wandern und neue gefäßähnliche Strukturen zu bilden nach, und sie erhöhten Moleküle, die vorbeiströmende Immunzellen festhalten – klassische Zeichen endotheler Dysfunktion.
Wie Gefäßzellen Makrophagen in einen schädlichen Zustand drängen
Um zu untersuchen, was das für das Verhalten von Immunzellen bedeutet, kultivierte das Team Ox‑LDL-behandelte Endothelzellen gemeinsam mit Makrophagen in einem Transwell-System, das den Austausch löslicher Signale erlaubt. Makrophagen, die gestressten Endothelzellen ausgesetzt waren, verlagerten sich deutlich in ein proinflammatorisches „M1“-Muster: Sie zeigten Oberflächenmarker, die mit Angriffszustand assoziiert sind, produzierten mehr entzündliche Substanzen wie TNF‑α und IL‑1β und wanderten verstärkt in Richtung der Endothelschicht. Als die Forschenden einen wichtigen Schalter innerhalb der Endothelzellen, den NF‑κB-Weg, blockierten, verringerten sich sowohl die Freisetzung von IL‑32γ als auch diese M1-Verschiebung. Auch die Neutralisierung von IL‑32γ selbst erzeugte einen ähnlichen Effekt, reduzierte Entzündungsmarker und dämpfte die Makrophagenbewegung – ein Hinweis darauf, dass IL‑32γ eine zentrale Vermittlerrolle spielt.
Ein Blick auf die Signalkette
Die Wissenschaftler fragten dann, ob IL‑32γ direkt auf Makrophagen wirken kann und über welchen Weg. Die Zugabe von gereinigtem IL‑32γ zu Makrophagen, ohne Endothelzellen, reichte aus, um sie in den M1-Zustand zu treiben und ihre Beweglichkeit zu erhöhen. Diese Reaktion hing von einer weiteren intrazellulären Signalkaskade ab, dem p38-MAPK-Weg: Wurde ein p38-blockierendes Medikament zugesetzt, konnte IL‑32γ die entzündlichen Gene, die Zytokinfreisetzung und die Migration nicht mehr steigern. 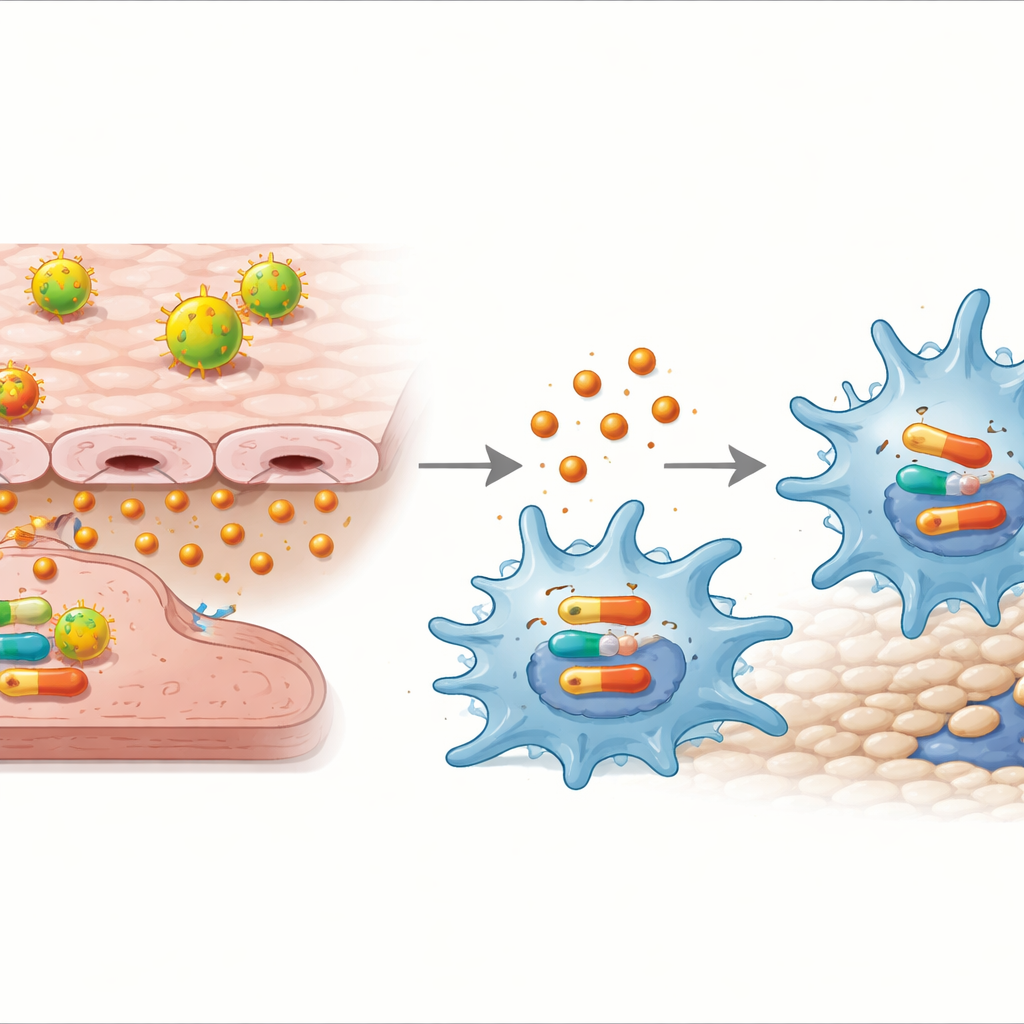
Was in lebenden Arterien passiert
Schließlich testete das Team den Weg in Mäusen, die genetisch anfällig für Atherosklerose sind und mit einer westlichen, fettreichen Diät gefüttert wurden. Mäuse, denen IL‑32γ injiziert wurde, entwickelten größere arterielle Plaques, zeigten mehr Makrophagenansammlungen in diesen Plaques und hatten höhere Blutspiegel entzündlicher Moleküle – trotz ähnlicher Cholesterinwerte im Vergleich zu Kontrollen. Wurde der p38-Weg in diesen Tieren blockiert, sanken Plaquegröße, Makrophagenansammlung und Entzündung wieder in Richtung Ausgangswerte. Das deutet darauf hin, dass IL‑32γ das Plaquewachstum in lebenden Arterien hauptsächlich durch die Verstärkung entzündlicher Signalwege beschleunigt und nicht durch eine Veränderung der Blutlipide.
Was das für zukünftige Therapien bedeutet
Insgesamt skizziert diese Arbeit eine Abfolge: Oxidiertes Cholesterin reizt die Gefäßauskleidung, diese Zellen reagieren mit der Freisetzung von IL‑32γ, und IL‑32γ treibt benachbarte Makrophagen über p38-Signale in einen aggressiven, plaquefördernden Zustand. Indem die Studie IL‑32γ als Schlüssellink zwischen geschädigten Endothelzellen und überaktiven Immunzellen identifiziert, schlägt sie eine neue therapeutische Richtung vor: Statt nur das Cholesterin zu senken, könnten künftige Medikamente auch IL‑32γ oder seinen p38-Weg anvisieren, um Entzündungen in der Gefäßwand zu dämpfen und das Fortschreiten der Atherosklerose zu verlangsamen oder möglicherweise umzukehren.
Zitation: Yang, Z., Liu, M., Shi, Y. et al. Upregulation of IL-32γ in endothelial cells promotes macrophage recruitment and inflammatory progression in atherosclerosis. Sci Rep 16, 11262 (2026). https://doi.org/10.1038/s41598-026-40151-z
Schlüsselwörter: Atherosklerose, Endothelzellen, Makrophagenpolarisation, Entzündung, IL-32 gamma