Clear Sky Science · tr
mist: tek hücre verilerinde diferansiyel DNA metilasyon dinamiklerini tespit etmek için hiyerarşik Bayesyen çerçeve
Hücrelerimizi Şekillendiren İşaretleri İzlemek
Vücudunuzdaki her hücre aynı DNA’yı taşır, ancak beyin hücreleri, kalp hücreleri ve bağışıklık hücreleri çok farklı davranır. Bunun bir nedeni, DNA üzerindeki metil grupları gibi kimyasal etiketlerin genleri açıp kapamaya yardımcı olmasıdır. Yeni araçlarla, bilim insanları artık bu etiketleri gelişen veya değişen binlerce tek hücrede okuyabiliyor. Bu yazı, bu devasa ve gürültülü ölçümleri gelişim ve hastalık sırasında bu DNA etiketlerinin zaman içinde nasıl kaydığına dair net hikâyelere dönüştüren istatistiksel bir yöntem olan “mist”i tanıtıyor.
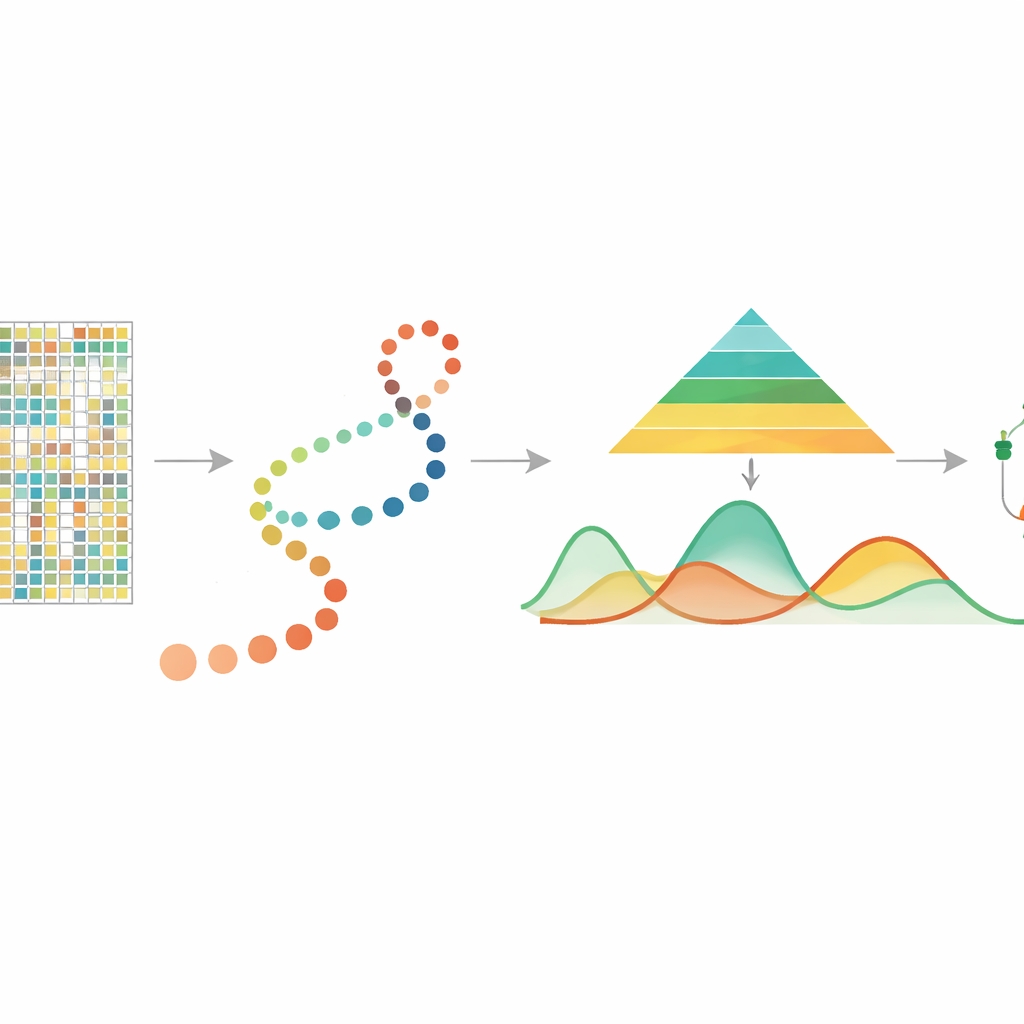
DNA Üzerindeki Kimyasal İpuçlarını Okumak
DNA metilasyonu, sıklıkla CpG adı verilen bölgelerde görülen sitozin bazına eklenen küçük bir kimyasal gruptur ve hangi genlerin aktif olduğunu kontrol etmede merkezî bir rol oynar. Toplu örneklerle yapılan önceki çalışmalar metilasyonun yaşlanma, stres tepkileri ve kanser ile ilişkili olduğunu ve hücre bölünmesi sırasında kalıtılabileceğini göstermiştir. Son zamanlarda, tek hücre DNA metilasyonu teknolojileri her bir hücre için metilasyonu ölçmeyi mümkün kılmış; bu da toplu örneklerde ortalaması alınarak kaybolacak zengin hücresel farklılıkları ortaya çıkardı. Ancak bu ölçümler seyrek ve gürültülüdür ve şimdiye kadar hücrelerin gelişimsel “sahte zaman” — hücreleri erken durumdan geçecek şekilde sıralayan çıkarımlı bir zaman çizelgesi — boyunca metilasyonun sürekli nasıl değiştiğini izlemek için özel bir araç yoktu.
Görünmez Bir Zaman Çizgisi Boyunca Hücreleri İzlemek
Birçok güncel deneyde araştırmacılar, gelişimsel veya hastalık sürecini temsil eden bir yol boyunca tek tek hücreleri düzenleyen başka yöntemlerle sahte zaman tahmini yapıyor. mist, başlangıç olarak bu hücre sıralamasını ve genler veya düzenleyici bölgeler (ör. promotörler) gibi gruplandırılmış tek hücre metilasyon verilerini alır. Ardından her genin metilasyon düzeyini sahte zaman üzerinde düzgün bir eğri olarak modelleyerek biyolojik değişkenlik miktarının erken ve geç aşamalar arasında farklı olmasına izin verir. Bu önemlidir çünkü erken gelişimsel aşamalar genellikle daha çeşitli ve esnektir, oysa daha geç aşamalar daha stabildir. Bu özellikleri hiyerarşik bir Bayesyen çerçeveye yerleştirerek mist, son derece seyrek verilerde rastgele gürültüden gerçek biyolojik desenleri ayırabilir.
Eğrilerden Önemli Genetik Oyunculara
mist her gen için düzgün bir metilasyon yörüngesi öğrendikten sonra önemli değişiklikleri bulmak için basit ama güçlü bir fikir kullanır: eğriler arasındaki alanı ölçer. Tek bir hücre grubunda, bir genin yörüngesini sabit bir düz çizgiyle karşılaştırarak sahte zaman boyunca güçlü değişiklik gösterenleri işaretler. İki grup, örneğin iki beyin bölgesi karşılaştırıldığında, iki yörüngeyi hizalar ve şekil farklarına odaklanarak genel olarak ne kadar ayrıldıklarını ölçer; sadece temel düzey farklılıklarına bakmaz. Kapsamlı bilgisayar simülasyonlarında mist, genelize edilebilen eklemeli modeller (GAM) ve standart polinom regresyon gibi yaygın alternatiflerden daha doğru şekilde gerçek alttaki metilasyon desenlerini geri kazandı ve diferansiyel metilasyon gösteren genleri tespit etti. Ayrıca sahte zamanı göz ardı eden başka bir metilasyon yöntemini de geride bırakarak hücrelerin zamansal sıralamasını açıkça modellemenin değerini vurguladı.
Epigenetik Gözlerle Gelişimi Görmek
Yazarlar, bu istatistiksel kazanımların biyolojik olarak ne anlama geldiğini göstermek için mist’i gerçek çok-omik veri setlerine uyguladılar. Fare embriyolarında mist, kalp gelişimini düzenleyenler, bağışıklık hücresi oluşumu ve kök hücre potansiyelinin kaybı gibi bilinen soy geçişlerini izleyen genlerin metilasyon desenlerini ortaya çıkardı. Gelişmekte olan insan beyin dokusunda ise, gestasyon döneminden yetişkinliğe kadar frontal korteks ve hipokampus arasında metilasyonun farklı şekilde evrildiği genleri açığa çıkardı. Örneğin, hafıza ile ilgili sinyalleşmede merkezi bir gen hipokampal yörünge boyunca azalan metilasyon gösterdi; bu da bu bölgede artan etkinlikle tutarlı. Öte yandan frontal kortekste büyümeyle ilişkili bir reseptör olgunlaştıkça daha çok metile oldu; bu da büyümeden uzun dönemli işleve geçişi düşündürüyor. Bu bulgular, mist’in DNA üzerindeki ince kimyasal değişiklikleri hücre kimliği ve beyin devrelerindeki büyük kaymalara bağlayabileceğini gösteriyor.
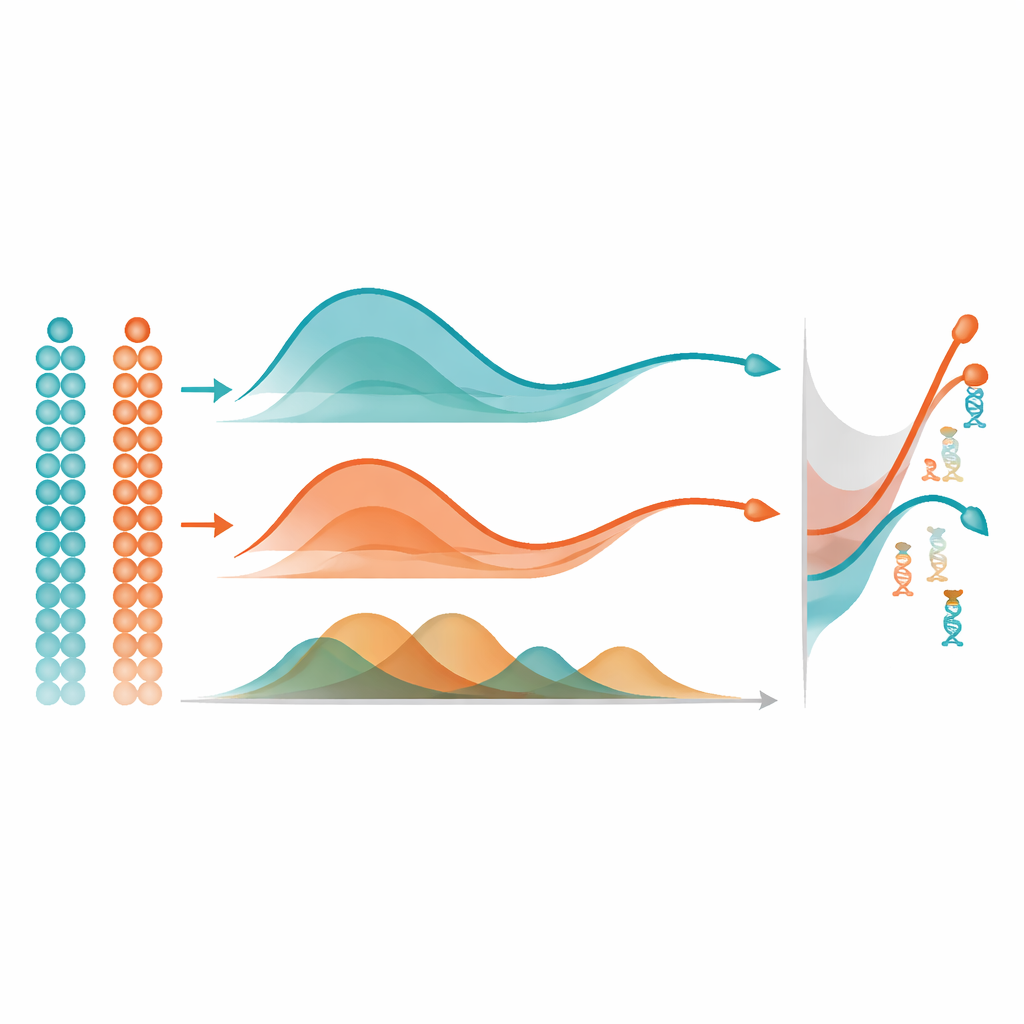
Gelecek Araştırmalar İçin Neden Önemli
Tek hücrelerde zaman içinde DNA metilasyon dinamiklerini takip etmek için ilkesel bir yol sunarak mist, epigenom çalışmalarının araç setinde önemli bir boşluğu dolduruyor. Yöntem özellikle seyrek, oransal metilasyon verileri için tasarlanmıştır ve hücreler gelişirken veya farklı soy hattına ya da doku bölgesine ayrılırken düzenleyici davranışı değişen genleri vurgulayabilir. Yöntem iyi sahte zaman tahminlerine bağlıdır ve çok ince genomik bölgeler için hesaplama açısından yoğun olabilir, ancak gen veya promotör düzeyinde zaten pratik olup açık kaynaklı yazılım olarak mevcuttur. Uzman olmayanlar için ana mesaj şudur: mist, devrimsel boyuttaki gürültülü tek hücre metilasyon veri setlerini gelişim, yaşlanma ve hastalık sırasında önemli düzenleyici anahtarların ne zaman ve nerede aktifleştirildiğine dair net haritalara dönüştürmeye yardımcı olur.
Atıf: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Anahtar kelimeler: tek hücre DNA metilasyonu, sahte zaman (pseudotime) analizi, Bayesyen modelleme, epigenetik düzenleme, gelişimsel yolculuklar