Clear Sky Science · de
mist: ein hierarchisches Bayessches Rahmenwerk zur Erkennung differentieller DNA-Methylierungsdynamiken in Einzelzell‑Daten
Den Markierungen folgen, die unsere Zellen formen
Jede Zelle in Ihrem Körper trägt dieselbe DNA, doch Nervenzellen, Herzmuskelzellen und Immunzellen verhalten sich sehr unterschiedlich. Ein Grund sind chemische Markierungen an der DNA, etwa Methylgruppen, die Gene ein- oder ausschalten helfen. Mit neuen Verfahren können Forschende diese Markierungen jetzt in Tausenden einzelner Zellen lesen, während sie sich entwickeln oder verändern. Dieser Artikel stellt „mist“ vor, eine statistische Methode, die diese riesigen, verrauschten Messungen in klare Erzählungen darüber verwandelt, wie sich DNA‑Markierungen im Verlauf von Entwicklung und Krankheit verändern.
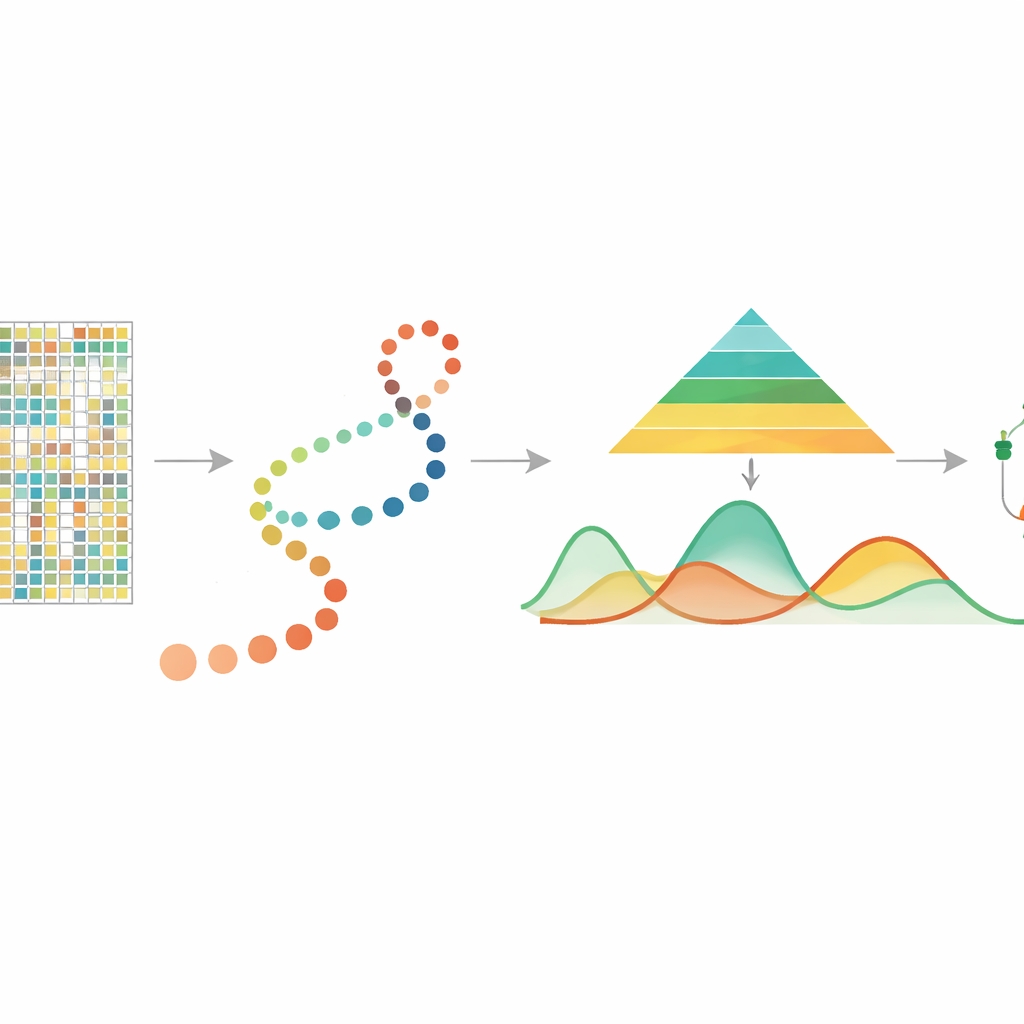
Chemische Hinweise auf der DNA lesen
DNA‑Methylierung ist eine kleine chemische Ergänzung an dem DNA‑Baustein Cytosin, häufig an Stellen, die CpGs genannt werden, und spielt eine zentrale Rolle bei der Steuerung, welche Gene aktiv sind. Frühere Studien an gepoolten (Bulk‑)Proben zeigten, dass Methylierung an Alterungsprozessen, Stressreaktionen und Krebs beteiligt ist und beim Zellteilen vererbt werden kann. Kürzlich erlauben Einzelzell‑Methylierungsverfahren, die Methylierung in jeder einzelnen Zelle zu messen und so reiche Unterschiede zwischen Zellen zu entdecken, die in Bulk‑Daten verwischt würden. Diese Messungen sind jedoch dünn besetzt und verrauscht, und bis jetzt gab es kein spezielles Werkzeug, um zu verfolgen, wie Methylierung kontinuierlich verändert wird, während Zellen einen entwicklungsbezogenen „Pseudotime“‑Verlauf durchlaufen — eine inferierte Zeitleiste, die Zellen von früheren zu späteren Zuständen ordnet.
Zellen entlang einer unsichtbaren Zeitleiste folgen
In vielen modernen Experimenten verwenden Forschende andere Methoden, um Pseudotime zu schätzen, und ordnen einzelne Zellen entlang eines Pfads, der einen Entwicklungs‑ oder Krankheitsprozess repräsentiert. mist nimmt diese Zellordnung zusammen mit Einzelzell‑Methylierungsdaten, gruppiert nach Genen oder Regionen wie Promotoren, als Ausgangspunkt. Die Methode modelliert dann das Methylierungsniveau jedes Gens als glatte Kurve über der Pseudotime und erlaubt, dass die biologische Variabilität zwischen frühen und späten Stadien unterschiedlich sein kann. Das ist wichtig, weil frühe Entwicklungsstadien oft vielfältiger und flexibler sind, während spätere Stadien stabiler werden. Indem diese Merkmale in ein hierarchisches bayessches Modell eingebaut werden, kann mist wahre biologische Muster von zufälligem Rauschen in hoch sparsamen Daten trennen.
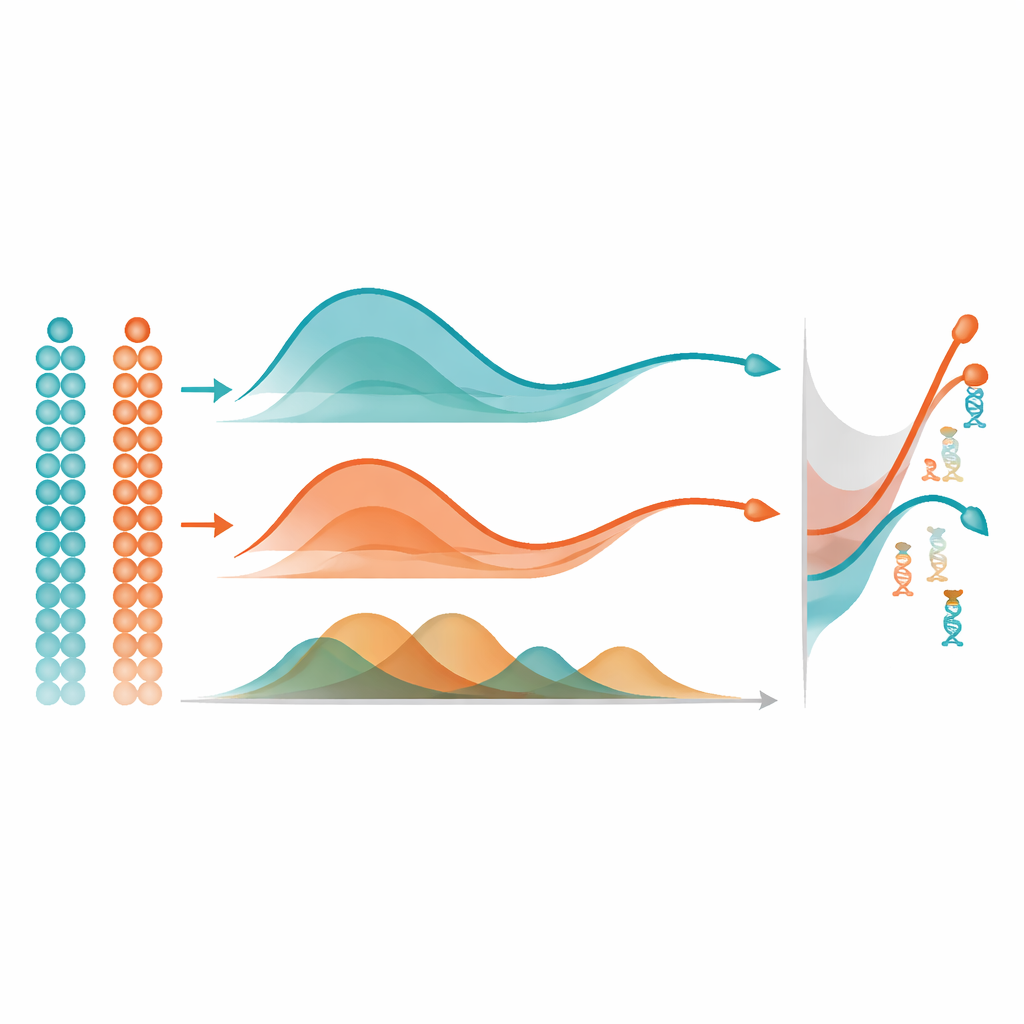
Von Kurven zu wichtigen genetischen Akteuren
Sobald mist für jedes Gen eine glatte Methylierungsentwicklung gelernt hat, nutzt es eine einfache, aber wirksame Idee, um wichtige Veränderungen zu finden: Es misst die Fläche zwischen Kurven. In einer einzelnen Zellgruppe vergleicht es die Trajektorie eines Gens mit einer Geraden, um Gene zu markieren, die sich stark über die Pseudotime verändern. Beim Vergleich zweier Gruppen, etwa zweier Gehirnregionen, gleicht es die beiden Trajektorien an und misst, wie weit sie insgesamt auseinanderliegen, wobei der Schwerpunkt auf Formunterschieden liegt und nicht nur auf dem Basisniveau. In umfangreichen Computersimulationen rekonstruierte mist die zugrundeliegenden Methylierungsmuster genauer und identifizierte differentielle methy lierte Gene besser als weit verbreitete Alternativen wie generalisierte additive Modelle und standardmäßige Polynomregression. Es übertraf auch eine andere Methylierungsmethode, die Pseudotime ignoriert, und unterstreicht damit den Wert, die zeitliche Ordnung der Zellen explizit zu modellieren.
Entwicklung durch epigenetische Augen sehen
Die Autorinnen und Autoren wandten mist auf reale Multi‑Omics‑Datensätze an, um zu zeigen, was diese statistischen Verbesserungen biologisch bedeuten. In Mausembryonen deckte mist Gene auf, deren Methylierungsmuster bekannte Linienübergänge widerspiegeln, einschließlich Regulatoren der Herzentwicklung, der Bildung von Immunzellen und des Verlusts von Stammzellpotenzial. In sich entwickelndem humanem Hirngewebe zeigte es Gene, deren Methylierung sich im frontalen Kortex und Hippocampus unterschiedlich entwickelte — über Stadien von der Gestation bis ins Erwachsenenalter. Beispielsweise zeigte ein zentrales Gen für gedächtnisbezogene Signalübertragung abnehmende Methylierung entlang der hippocampalen Trajektorie, konsistent mit zunehmender Aktivität in dieser Region, während ein wachstumsbezogener Rezeptor im frontalen Kortex mit der Reifung des Gewebes stärker methyliert wurde, was auf eine Verschiebung von Wachstum hin zu langfristiger Funktion hindeutet. Diese Befunde veranschaulichen, wie mist subtile chemische Veränderungen an der DNA mit großen Verschiebungen in Zellidentität und neuronaler Verschaltung verbinden kann.
Warum das für künftige Forschung wichtig ist
Indem mist eine prinzipienbasierte Möglichkeit bietet, DNA‑Methylierungsdynamiken in Einzelzellen über die Zeit zu verfolgen, schließt es eine wichtige Lücke im Werkzeugkasten zur Erforschung des Epigenoms. Es ist speziell für sparsame, proportionale Methylierungsdaten konzipiert und kann Gene hervorheben, deren regulatorisches Verhalten sich verändert, während Zellen sich entwickeln oder in unterschiedliche Linien oder Gewebeareale aufspalten. Obwohl die Methode auf guten Pseudotime‑Schätzungen beruht und bei sehr feinen genomischen Regionen rechenintensiv sein kann, ist sie bereits auf Gen‑ oder Promotor‑Ebene praktisch anwendbar und als Open‑Source‑Software verfügbar. Für Nicht‑Spezialistinnen und Nicht‑Spezialisten ist die Hauptbotschaft, dass mist dabei hilft, riesige, verrauschte Einzelzell‑Methylierungsdatensätze in klare Karten zu übersetzen, die zeigen, wann und wo wichtige regulatorische Schalter während Entwicklung, Alterung und Krankheit umgelegt werden.
Zitation: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Schlüsselwörter: Einzelzell‑DNA‑Methylierung, Pseudotime‑Analyse, Bayessche Modellierung, epigenetische Regulation, entwicklungsbedingte Pfade