Clear Sky Science · pt
mist: uma estrutura bayesiana hierárquica para detectar dinâmicas diferenciais de metilação do DNA em dados de célula única
Seguindo as Marcas que Moldam Nossas Células
Cada célula do seu corpo carrega o mesmo DNA, ainda que células cerebrais, cardíacas e imunológicas se comportem de maneira muito diferente. Uma razão são as marcas químicas no DNA, como grupos metil, que ajudam a ligar ou desligar genes. Com novas ferramentas, cientistas podem hoje ler essas marcas em milhares de células individuais enquanto elas se desenvolvem ou mudam. Este artigo apresenta o “mist”, um método estatístico que transforma essas medições massivas e ruidosas em narrativas claras sobre como essas marcas de DNA mudam ao longo do tempo no desenvolvimento e na doença.
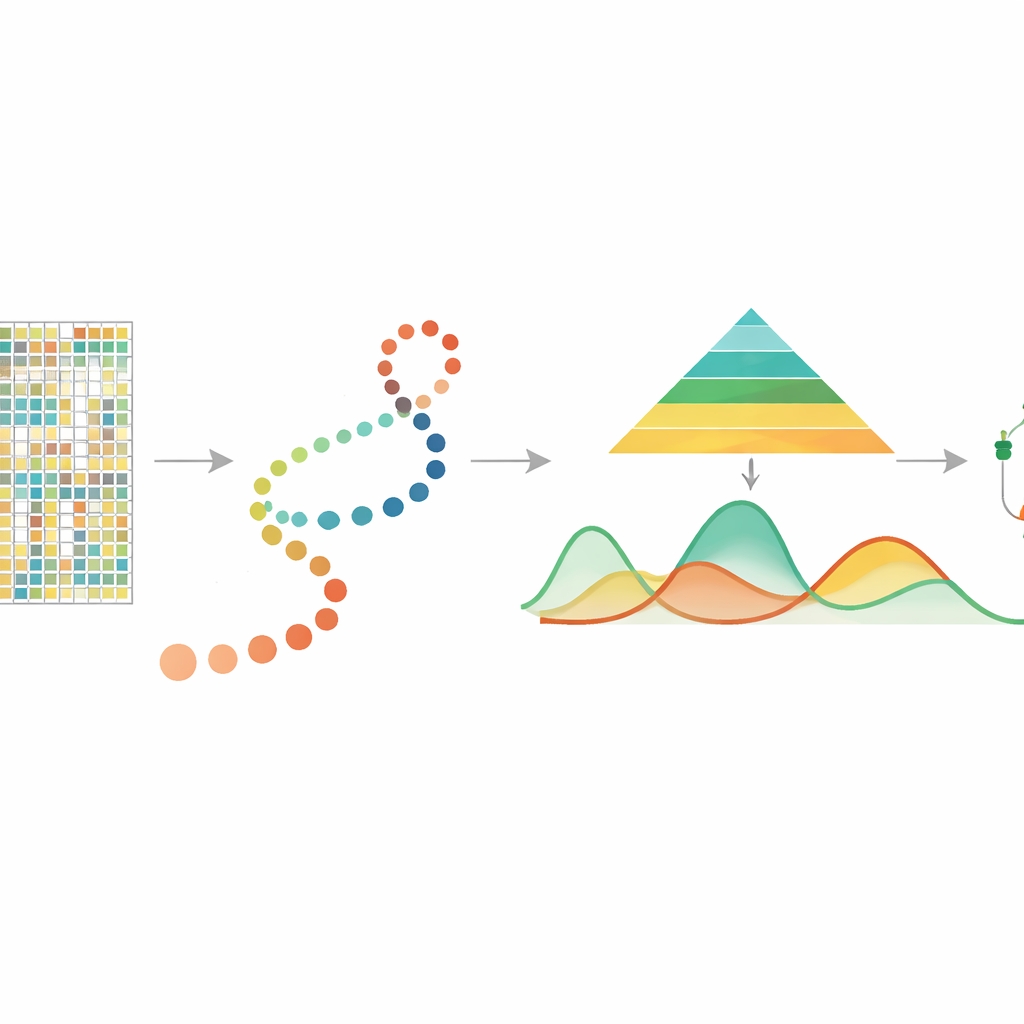
Lendo Pistas Químicas no DNA
A metilação do DNA é uma pequena adição química à base citosina, frequentemente em sítios chamados CpG, e desempenha papel central no controle de quais genes estão ativos. Estudos anteriores com amostras em conjunto mostraram que a metilação está envolvida no envelhecimento, nas respostas ao estresse e no câncer, e que pode ser herdada quando as células se dividem. Recentemente, tecnologias de metilação do DNA em célula única possibilitaram medir a metilação em cada célula individual, revelando diferenças ricas entre células que seriam suavizadas em análises de conjunto. Contudo, essas medições são esparsas e ruidosas, e até agora não havia uma ferramenta dedicada para acompanhar como a metilação muda continuamente à medida que as células progridem em um “pseudotempo” de desenvolvimento — uma linha do tempo inferida que ordena as células de estados mais iniciais a mais tardios.
Seguindo Células ao Longo de uma Linha do Tempo Invisível
Em muitos experimentos modernos, pesquisadores usam outros métodos para estimar o pseudotempo, organizando células individuais ao longo de um caminho que representa um processo de desenvolvimento ou doença. O mist usa essa ordenação celular, juntamente com dados de metilação do DNA em célula única agrupados por genes ou regiões como promotores, como ponto de partida. Em seguida, modela o nível de metilação de cada gene como uma curva suave ao longo do pseudotempo, permitindo que a quantidade de variabilidade biológica difira entre estágios iniciais e tardios. Isso é importante porque estágios iniciais do desenvolvimento costumam ser mais diversos e flexíveis, enquanto estágios tardios são mais estáveis. Ao incorporar essas características em uma estrutura bayesiana hierárquica, o mist consegue separar padrões biológicos verdadeiros do ruído aleatório em dados altamente esparsos.
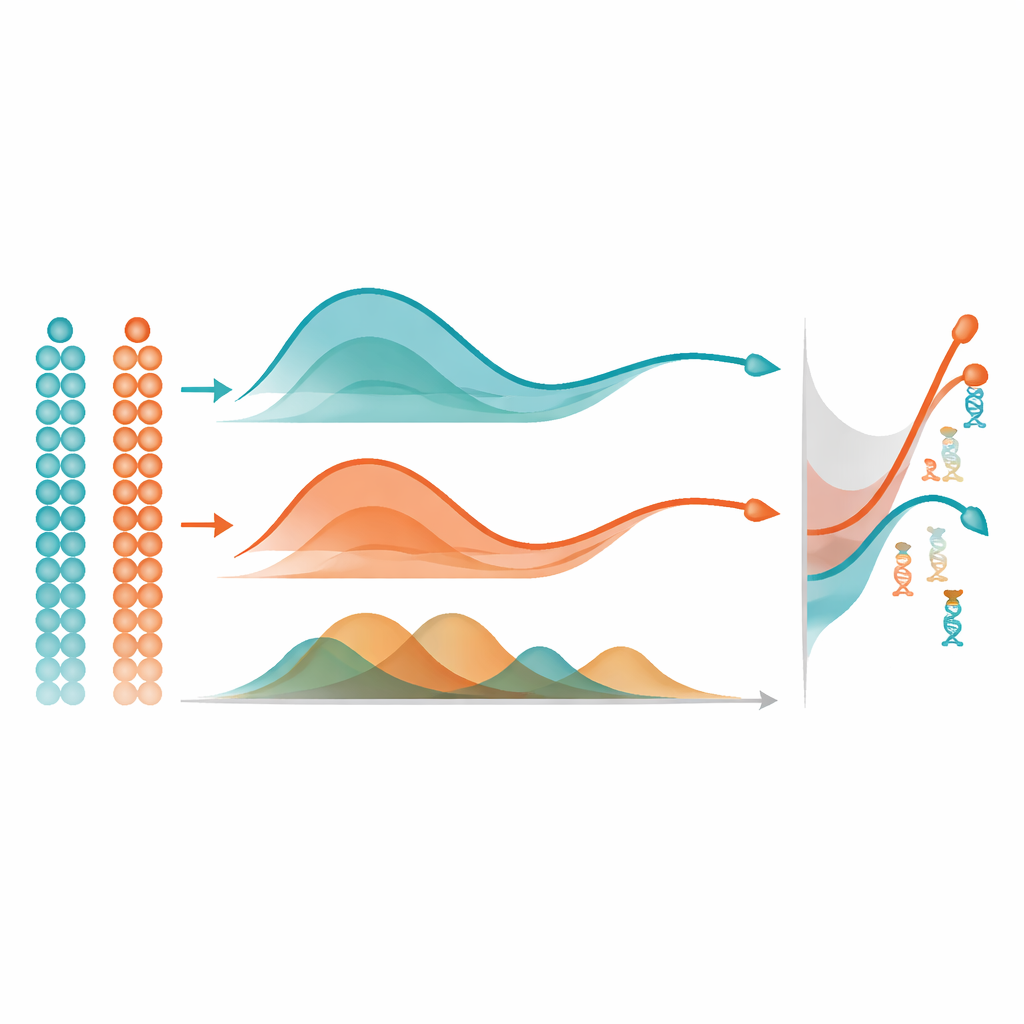
De Curvas a Jogadores Genéticos Chave
Uma vez que o mist aprendeu uma trajetória suave de metilação para cada gene, ele usa uma ideia simples porém poderosa para encontrar mudanças importantes: mede a área entre curvas. Em um único grupo de células, compara a trajetória de um gene com uma linha plana para sinalizar aqueles que mudam fortemente ao longo do pseudotempo. Ao comparar dois grupos, como duas regiões cerebrais, alinha as duas trajetórias e mede quão distantes elas estão no conjunto, focando em diferenças de forma em vez de apenas no nível basal. Em extensas simulações computacionais, o mist recuperou de forma mais precisa os verdadeiros padrões subjacentes de metilação e identificou genes diferencialmente metilados com mais acurácia do que alternativas amplamente usadas, como modelos aditivos generalizados e regressão polinomial padrão. Também superou outro método de metilação que ignora o pseudotempo, destacando o valor de modelar explicitamente a ordenação temporal das células.
Vendo o Desenvolvimento pelos Olhos da Epigenética
Os autores aplicaram o mist a conjuntos de dados multi-ômicos reais para mostrar o que esses ganhos estatísticos significam biologicamente. Em embriões de camundongo, o mist revelou genes cujos padrões de metilação acompanharam transições de linhagem conhecidas, incluindo reguladores do desenvolvimento cardíaco, formação de células imunes e perda do potencial de células-tronco. Em tecido cerebral humano em desenvolvimento, mostrou genes cuja metilação evoluiu de forma diferente no córtex frontal e no hipocampo em estágios desde a gestação até a idade adulta. Por exemplo, um gene central para sinalização relacionada à memória apresentou diminuição da metilação ao longo da trajetória hipocampal, consistente com aumento da atividade nessa região, enquanto um receptor associado ao crescimento no córtex frontal tornou-se mais metilado à medida que o tecido amadurecia, sugerindo uma transição do crescimento para funções de longo prazo. Essas descobertas ilustram como o mist pode conectar mudanças químicas sutis no DNA a grandes alterações na identidade celular e na circuitaria cerebral.
Por Que Isso Importa para Pesquisas Futuras
Ao oferecer uma forma fundamentada de acompanhar dinâmicas de metilação do DNA em células únicas ao longo do tempo, o mist preenche uma lacuna importante no conjunto de ferramentas para estudar o epigenoma. Ele foi projetado especificamente para dados de metilação proporcionais e esparsos, e pode destacar genes cujo comportamento regulatório muda conforme as células se desenvolvem ou divergem em diferentes linhagens ou regiões teciduais. Embora o método dependa de boas estimativas de pseudotempo e possa ser computacionalmente intensivo para regiões genômicas muito finas, já é prático ao nível de gene ou promotor e está disponível como software de código aberto. Para não especialistas, a mensagem principal é que o mist ajuda a traduzir enormes e ruidosos conjuntos de dados de metilação em célula única em mapas claros de quando e onde interruptores regulatórios importantes são acionados durante o desenvolvimento, envelhecimento e doença.
Citação: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Palavras-chave: metilação do DNA em célula única, análise de pseudotempo, modelagem Bayesiana, regulação epigenética, trajetórias do desenvolvimento