Clear Sky Science · nl
mist: een hiërarchisch Bayesiaans kader voor het detecteren van differentiële DNA-methyleringsdynamiek in single-cellgegevens
De sporen volgen die onze cellen vormen
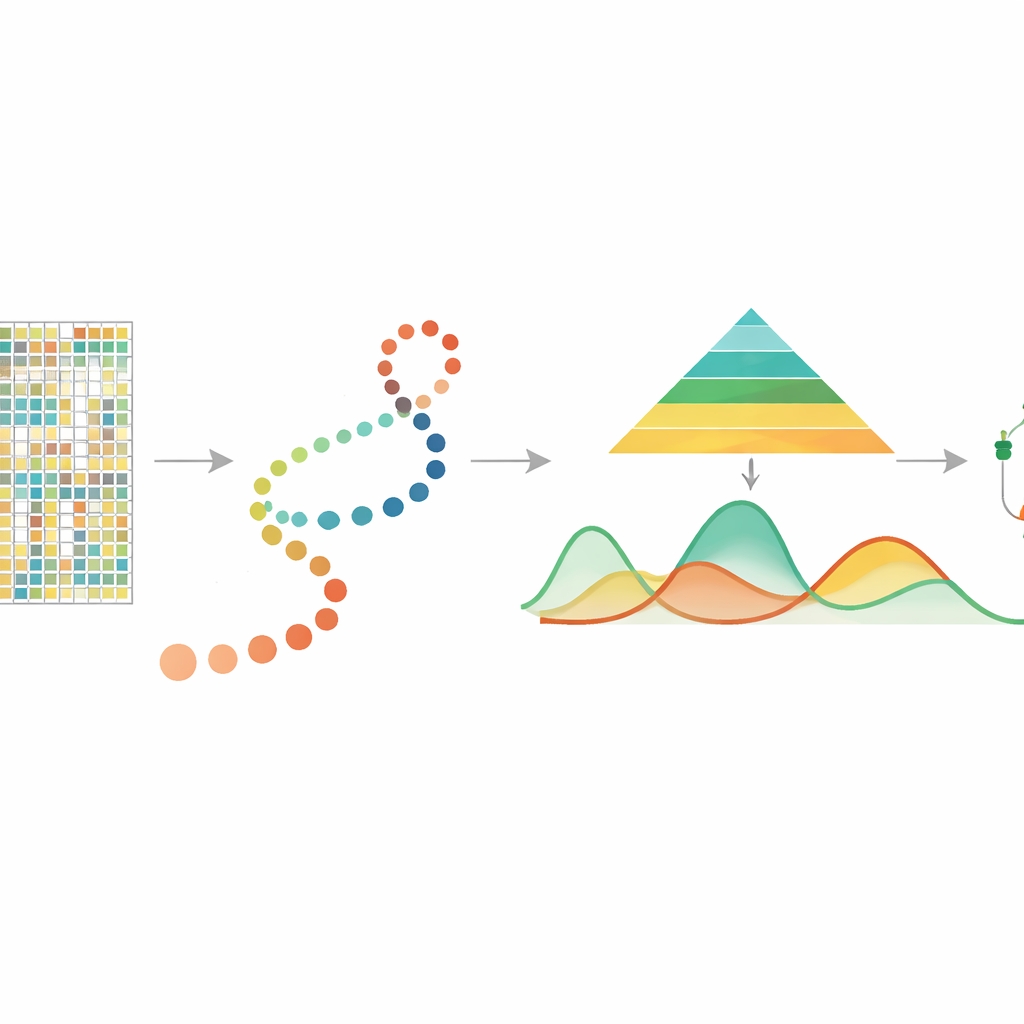
Elke cel in je lichaam draagt hetzelfde DNA, maar zenuwcellen, hartcellen en immuuncellen gedragen zich heel verschillend. Een oorzaak zijn chemische labels op het DNA, zoals methylgroepen, die helpen genen aan- of uit te zetten. Met nieuwe technieken kunnen wetenschappers deze labels nu in duizenden individuele cellen aflezen terwijl ze zich ontwikkelen of veranderen. Dit artikel introduceert “mist”, een statistische methode die die enorme, ruisige metingen omzet in duidelijke verhalen over hoe deze DNA-labels in de loop van de tijd veranderen bij ontwikkeling en ziekte.
Chemische aanwijzingen op DNA lezen
DNA-methylering is een kleine chemische toevoeging aan de DNA-basiscytosine, vaak op plaatsen die CpG's worden genoemd, en speelt een centrale rol in het regelen welke genen actief zijn. Eerdere studies met bulksamples toonden aan dat methylering betrokken is bij veroudering, stressreacties en kanker, en dat ze bij celdeling kan worden doorgegeven. Recentelijk maken single-cell DNA-methyleringstechnieken het mogelijk methylering per individuele cel te meten, waardoor rijke verschillen tussen cellen zichtbaar worden die in bulk zouden worden gemiddeld. Deze metingen zijn echter zeldzaam en ruisend, en tot nu toe ontbrak een speciaal hulpmiddel om te volgen hoe methylering continu verandert terwijl cellen door een ontwikkelings-“pseudotijd” gaan — een afgeleide tijdlijn die cellen ordent van vroegere naar latere toestanden.
Cellen volgen langs een onzichtbare tijdlijn
In veel moderne experimenten gebruiken onderzoekers andere methoden om pseudotijd te schatten, en rangschikken ze individuele cellen langs een pad dat een ontwikkelings- of ziekteproces vertegenwoordigt. mist neemt deze celordening, samen met single-cell DNA-methyleringsgegevens gegroepeerd per gen of regio’s zoals promoters, als uitgangspunt. Het modelleert vervolgens het methyleringsniveau van elk gen als een vloeiende kromme over pseudotijd, waarbij het toestaat dat de hoeveelheid biologische variabiliteit verschilt tussen vroege en late stadia. Dat is belangrijk omdat vroege ontwikkelingsstadia vaak diverser en flexibeler zijn, terwijl latere stadia stabieler zijn. Door deze kenmerken in een hiërarchisch Bayesiaans kader op te nemen, kan mist echte biologische patronen scheiden van willekeurige ruis in sterk sparse data.
Van krommen naar belangrijke genetische spelers
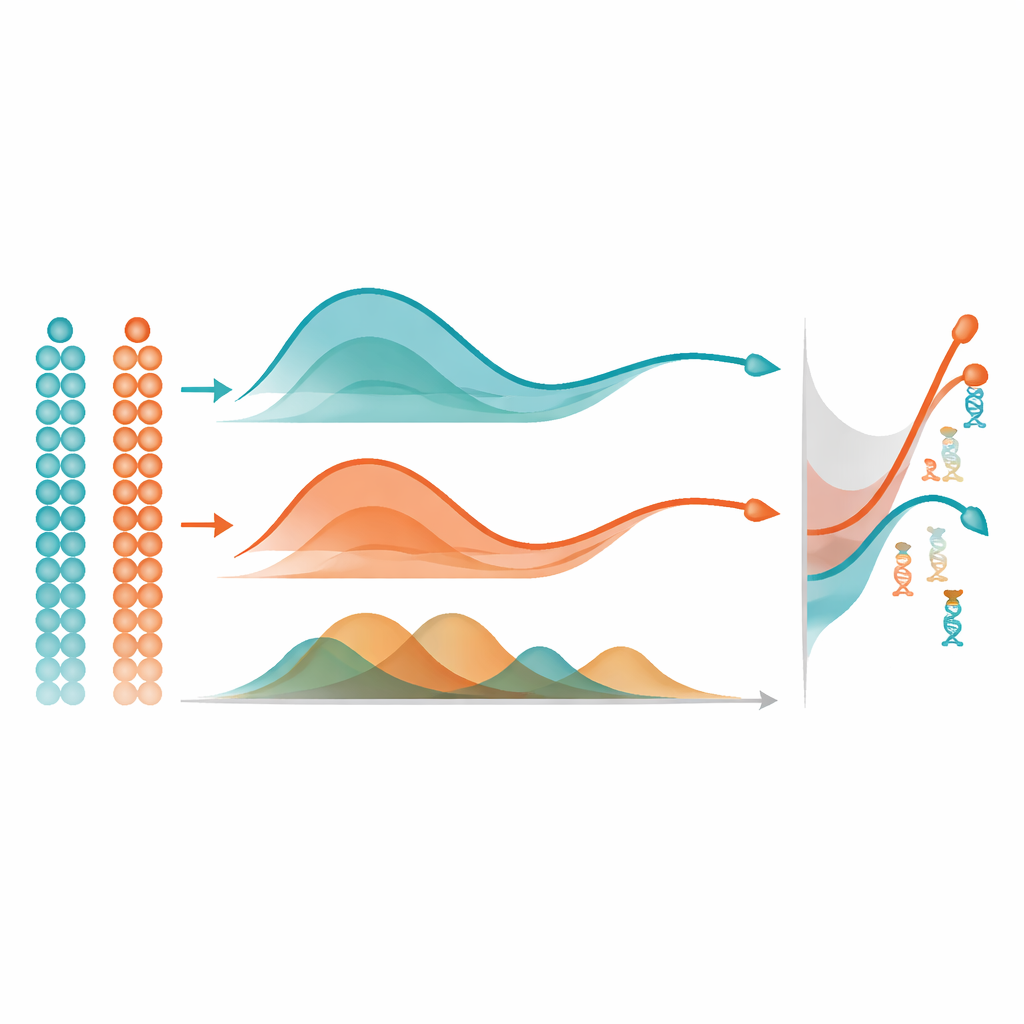
Zodra mist voor elk gen een vloeiende methyleringstraject heeft geleerd, gebruikt het een eenvoudig maar krachtig idee om belangrijke veranderingen te vinden: het meet de oppervlakte tussen krommen. Binnen één groep cellen vergelijkt het het traject van een gen met een vlakke lijn om diegenen te signaleren die sterk veranderen over pseudotijd. Bij vergelijking van twee groepen, bijvoorbeeld twee hersengebieden, lijnt het de twee trajecten op elkaar uit en meet het hoe ver ze in het algemeen uit elkaar liggen, met nadruk op verschillen in vorm in plaats van alleen het basisniveau. In uitgebreide computersimulaties reconstructeerde mist de onderliggende methyleringspatronen en identificeerde differentieel gemethyleerde genen nauwkeuriger dan veelgebruikte alternatieven zoals gegeneraliseerde additieve modellen en standaard polynoomregressie. Het presteerde ook beter dan een andere methyleringsmethode die pseudotijd negeert, wat de waarde benadrukt van expliciet modelleren van de temporele ordening van cellen.
Ontwikkeling zien door epigenetische ogen
De auteurs pasten mist toe op echte multi-omics datasets om te laten zien wat deze statistische verbeteringen biologisch betekenen. In muizembryo's bracht mist genen aan het licht waarvan de methyleringspatronen bekende lijntransities volgden, inclusief regulatoren van hartontwikkeling, vorming van immuuncellen en het verlies van stamcelpotentieel. In zich ontwikkelend menselijk hersenweefsel onthulde het genen waarvan de methylering zich anders ontwikkelde in de frontale cortex en hippocampus over stadia van zwangerschap tot volwassenheid. Bijvoorbeeld, een gen dat centraal staat in geheugengerelateerde signaaloverdracht toonde afnemende methylering langs het hippocampale traject, consistent met toenemende activiteit in dit gebied, terwijl een groeigerelateerd receptor in de frontale cortex meer gemethyleerd werd naarmate het weefsel rijpte, wat wijst op een verschuiving van groei naar langdurige functie. Deze bevindingen illustreren hoe mist subtiele chemische veranderingen op DNA kan koppelen aan grote verschuivingen in celidentiteit en hersencircuits.
Waarom dit belangrijk is voor toekomstig onderzoek
Door een principiële manier te bieden om DNA-methyleringsdynamiek in single cells over tijd te volgen, vult mist een belangrijke leemte in de gereedschapskist voor het bestuderen van het epigenoom. Het is speciaal ontworpen voor sparse, proportionele methyleringsdata en kan genen aanwijzen waarvan het regulatoire gedrag verandert terwijl cellen zich ontwikkelen of uiteenlopen in verschillende lijnages of weefselregio's. Hoewel de methode afhankelijk is van goede pseudotijdschattingen en bij zeer fijne genomische regio's rekenintensief kan zijn, is het al praktisch op gen- of promotorniveau en beschikbaar als open-source software. Voor niet-specialisten is de kernboodschap dat mist helpt enorme, ruisige single-cell methyleringsdatasets om te zetten in heldere kaarten van wanneer en waar belangrijke regulerende schakelaars worden omgezet tijdens ontwikkeling, veroudering en ziekte.
Bronvermelding: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Trefwoorden: single-cell DNA-methylering, pseudotime-analyse, Bayesiaanse modellering, epigenetische regulatie, ontwikkelingsbanen