Clear Sky Science · es
mist: un marco bayesiano jerárquico para detectar dinámicas diferenciales de metilación del ADN en datos de célula única
Siguiendo las marcas que moldean nuestras células
Cada célula de tu cuerpo contiene el mismo ADN, sin embargo las neuronas, las células cardíacas y las células inmunitarias se comportan de forma muy diferente. Una razón son las etiquetas químicas en el ADN, como los grupos metilo, que ayudan a activar o silenciar genes. Con nuevas herramientas, los científicos pueden ahora leer estas etiquetas en miles de células individuales a medida que se desarrollan o cambian. Este artículo presenta “mist”, un método estadístico que convierte esas mediciones masivas y ruidosas en relatos claros sobre cómo estas marcas del ADN cambian con el tiempo en el desarrollo y la enfermedad.
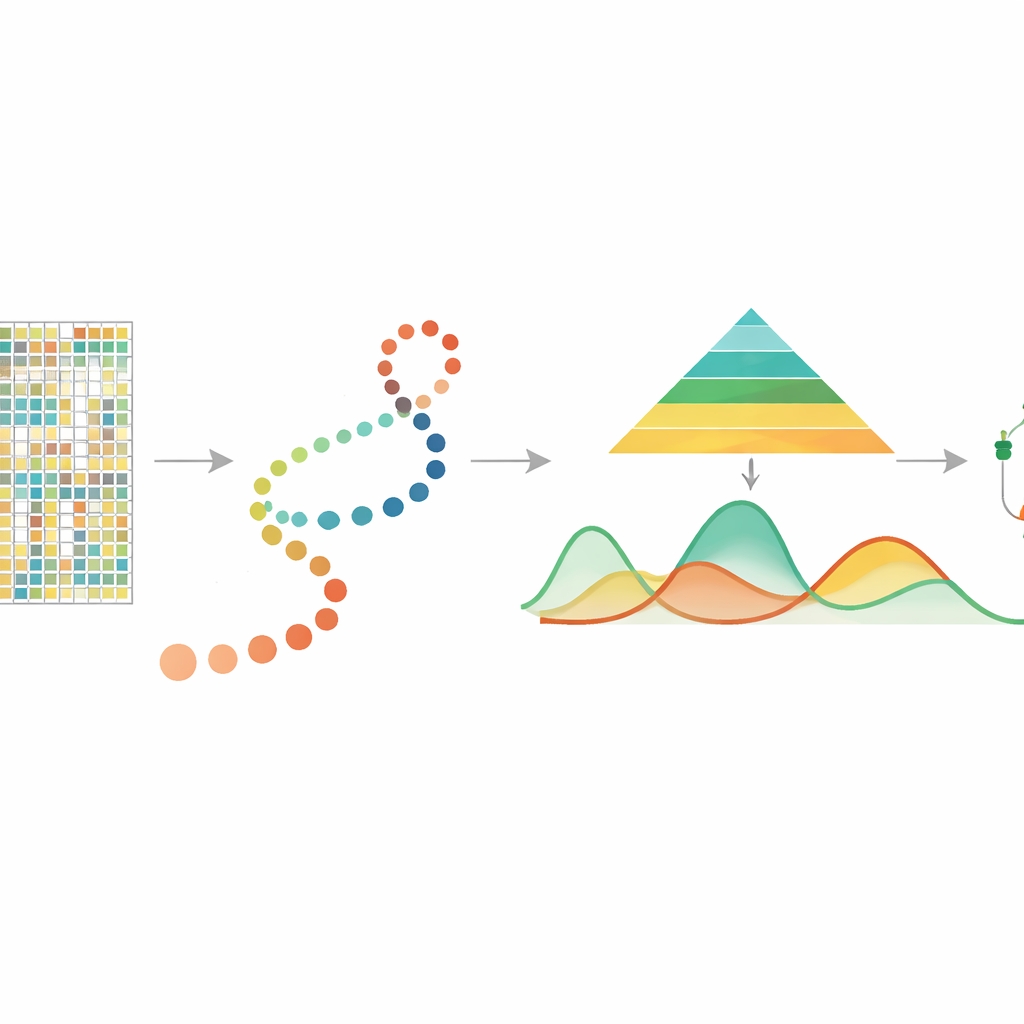
Leyendo pistas químicas en el ADN
La metilación del ADN es una pequeña adición química a la letra citosina del ADN, a menudo en sitios llamados CpG, y desempeña un papel central en el control de qué genes están activos. Estudios previos con muestras agregadas mostraron que la metilación está implicada en el envejecimiento, las respuestas al estrés y el cáncer, y que puede heredarse cuando las células se dividen. Recientemente, las tecnologías de metilación del ADN a nivel de célula única han hecho posible medir la metilación en cada célula individual, revelando ricas diferencias entre células que se perderían en muestras agregadas. Sin embargo, estas mediciones son escasas y ruidosas, y hasta ahora no existía una herramienta dedicada para seguir cómo la metilación cambia de forma continua a medida que las células avanzan a lo largo del “pseudotiempo” del desarrollo — una línea temporal inferida que ordena las células de estados tempranos a tardíos.
Siguiendo las células a lo largo de una línea temporal invisible
En muchos experimentos modernos, los investigadores usan otros métodos para estimar el pseudotiempo, disponiendo las células individuales a lo largo de un camino que representa un proceso de desarrollo o enfermedad. mist toma este ordenamiento celular, junto con datos de metilación del ADN de célula única agrupados por genes o regiones como promotores, como punto de partida. A continuación modela el nivel de metilación de cada gen como una curva suave sobre el pseudotiempo, permitiendo que la cantidad de variabilidad biológica difiera entre etapas tempranas y tardías. Esto es importante porque las etapas tempranas del desarrollo suelen ser más diversas y flexibles, mientras que las etapas tardías son más estables. Al incorporar estas características en un marco bayesiano jerárquico, mist puede separar patrones biológicos reales del ruido aleatorio en datos altamente escasos.
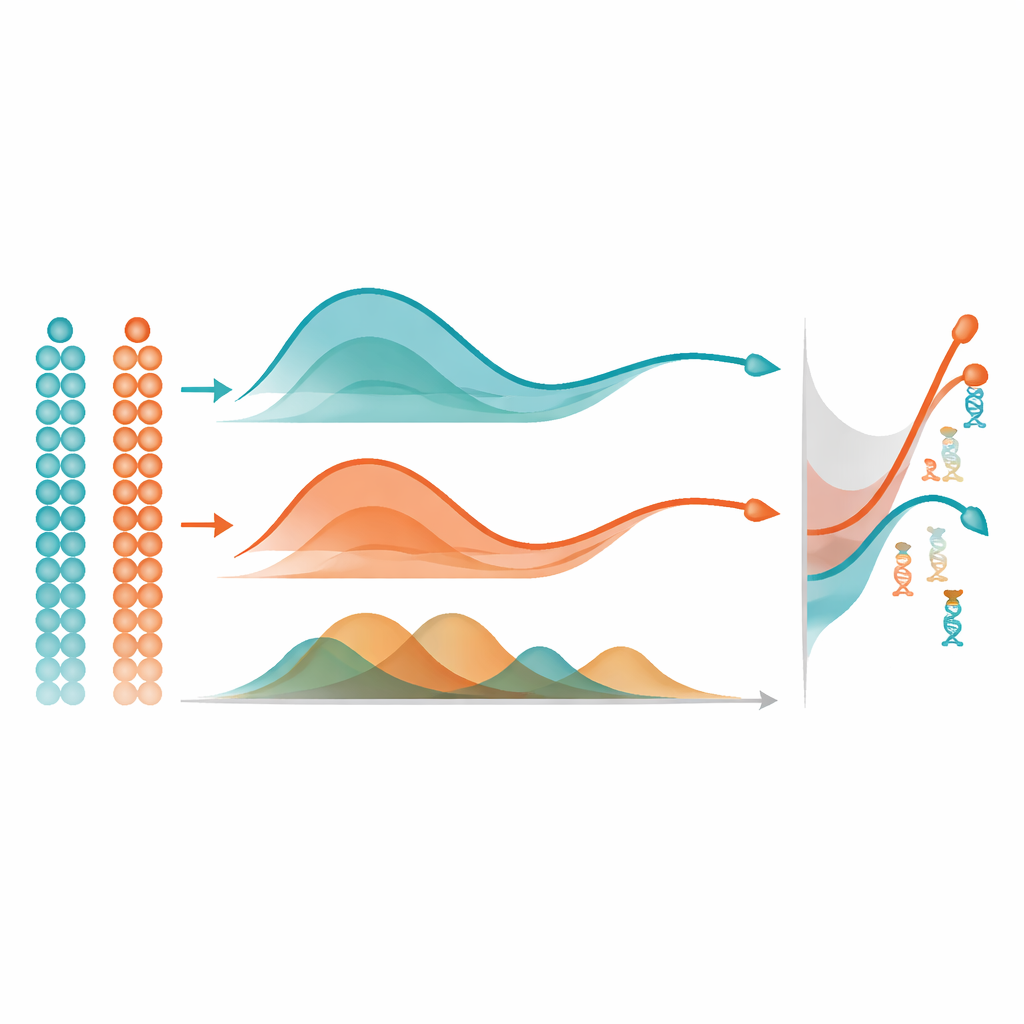
De curvas a actores genéticos clave
Una vez que mist ha aprendido una trayectoria suave de metilación para cada gen, utiliza una idea sencilla pero poderosa para encontrar cambios importantes: mide el área entre curvas. En un solo grupo de células, compara la trayectoria de un gen con una línea plana para señalar aquellos que cambian de forma marcada a lo largo del pseudotiempo. Al comparar dos grupos, como dos regiones cerebrales, alinea las dos trayectorias y mide cuánto se separan en conjunto, centrándose en diferencias de forma más que en diferencias de nivel basal. En extensas simulaciones por ordenador, mist recuperó con mayor precisión los patrones verdaderos de metilación subyacentes e identificó genes diferencialmente metilados mejor que alternativas de uso general como modelos aditivos generalizados y regresión polinómica estándar. También superó a otro método de metilación que ignora el pseudotiempo, destacando el valor de modelar explícitamente el orden temporal de las células.
Ver el desarrollo a través de ojos epigenéticos
Los autores aplicaron mist a conjuntos de datos multi-ómicos reales para mostrar qué significan estas mejoras estadísticas a nivel biológico. En embriones de ratón, mist descubrió genes cuyas pautas de metilación siguieron transiciones de linaje conocidas, incluidos reguladores del desarrollo cardíaco, la formación de células inmunitarias y la pérdida del potencial de células madre. En tejido cerebral humano en desarrollo, reveló genes cuya metilación evolucionó de forma diferente en la corteza frontal y el hipocampo a lo largo de etapas desde la gestación hasta la edad adulta. Por ejemplo, un gen central para la señalización relacionada con la memoria mostró una metilación decreciente a lo largo de la trayectoria hipocampal, coherente con una mayor actividad en esta región, mientras que un receptor relacionado con el crecimiento en la corteza frontal se volvió más metilado a medida que el tejido maduraba, lo que sugiere un cambio del crecimiento hacia la función a largo plazo. Estos hallazgos ilustran cómo mist puede conectar cambios químicos sutiles en el ADN con cambios importantes en la identidad celular y el cableado cerebral.
Por qué esto importa para la investigación futura
Al ofrecer una forma fundamentada de seguir las dinámicas de metilación del ADN en células individuales a lo largo del tiempo, mist llena una laguna clave en el conjunto de herramientas para estudiar el epigenoma. Está diseñado específicamente para datos de metilación proporcionales y escasos y puede señalar genes cuyo comportamiento regulador cambia a medida que las células se desarrollan o se diversifican en diferentes linajes o regiones tisulares. Aunque el método depende de buenas estimaciones de pseudotiempo y puede ser intensivo en cómputo para regiones genómicas muy finas, ya es práctico a nivel de gen o promotor y está disponible como software de código abierto. Para los no especialistas, el mensaje principal es que mist ayuda a traducir enormes y ruidosos conjuntos de datos de metilación de célula única en mapas claros de cuándo y dónde se activan interruptores regulatorios importantes durante el desarrollo, el envejecimiento y la enfermedad.
Cita: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Palabras clave: metilación del ADN en célula única, análisis de pseudotiempo, modelado bayesiano, regulación epigenética, trayectorias del desarrollo