Clear Sky Science · it
mist: un quadro bayesiano gerarchico per rilevare dinamiche differenziali della metilazione del DNA in dati single-cell
Seguire i marchi che plasmano le nostre cellule
Ogni cellula del tuo corpo contiene lo stesso DNA, eppure le cellule cerebrali, cardiache e del sistema immunitario si comportano in modo molto diverso. Una ragione sono i segnali chimici sul DNA, come i gruppi metilici, che aiutano ad accendere o spegnere i geni. Con nuovi strumenti, gli scienziati possono ora leggere questi segnali in migliaia di singole cellule mentre si sviluppano o cambiano. Questo articolo presenta “mist”, un metodo statistico che trasforma quelle misurazioni massive e rumorose in storie chiare su come questi marchi del DNA cambiano nel tempo durante lo sviluppo e la malattia.
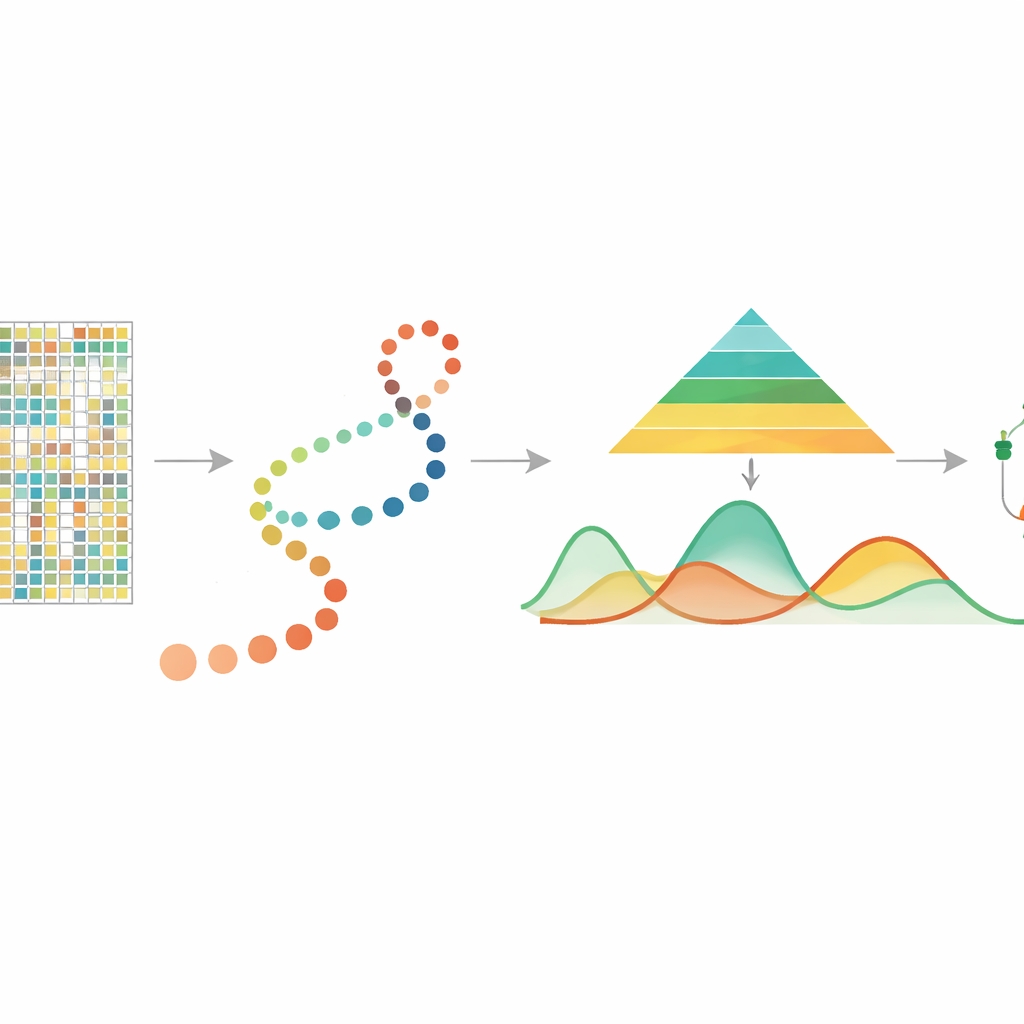
Leggere gli indizi chimici sul DNA
La metilazione del DNA è una piccola aggiunta chimica alla base citosina del DNA, spesso in siti detti CpG, e svolge un ruolo centrale nel controllare quali geni sono attivi. Studi precedenti su campioni bulk hanno mostrato che la metilazione è coinvolta nell’invecchiamento, nelle risposte allo stress e nel cancro, e che può essere trasmessa durante la divisione cellulare. Recentemente, le tecnologie di metilazione del DNA a singola cellula hanno reso possibile misurare la metilazione di ogni cellula individuale, rivelando ricche differenze tra cellule che verrebbero mediate in campioni bulk. Tuttavia, queste misurazioni sono scarse e rumorose, e fino ad ora non esisteva uno strumento dedicato per seguire come la metilazione cambi continuamente mentre le cellule progrediscono lungo un “pseudotempo” — una timeline inferita che ordina le cellule da stati più precoci a più tardivi.
Seguire le cellule lungo una timeline invisibile
In molti esperimenti moderni, i ricercatori usano altri metodi per stimare il pseudotempo, disponendo le singole cellule lungo un percorso che rappresenta un processo di sviluppo o patologico. mist prende questo ordinamento cellulare, insieme ai dati di metilazione del DNA single-cell raggruppati per geni o regioni come i promotori, come punto di partenza. Modella quindi il livello di metilazione di ciascun gene come una curva liscia nel corso del pseudotempo, permettendo che la quantità di variabilità biologica differisca tra stadi precoci e tardivi. Questo è importante perché gli stadi precoci dello sviluppo sono spesso più diversi e flessibili, mentre gli stadi successivi sono più stabili. Integrando queste caratteristiche in un quadro bayesiano gerarchico, mist riesce a separare i veri schemi biologici dal rumore casuale in dati altamente scarsi.
Dalle curve ai giocatori genetici chiave
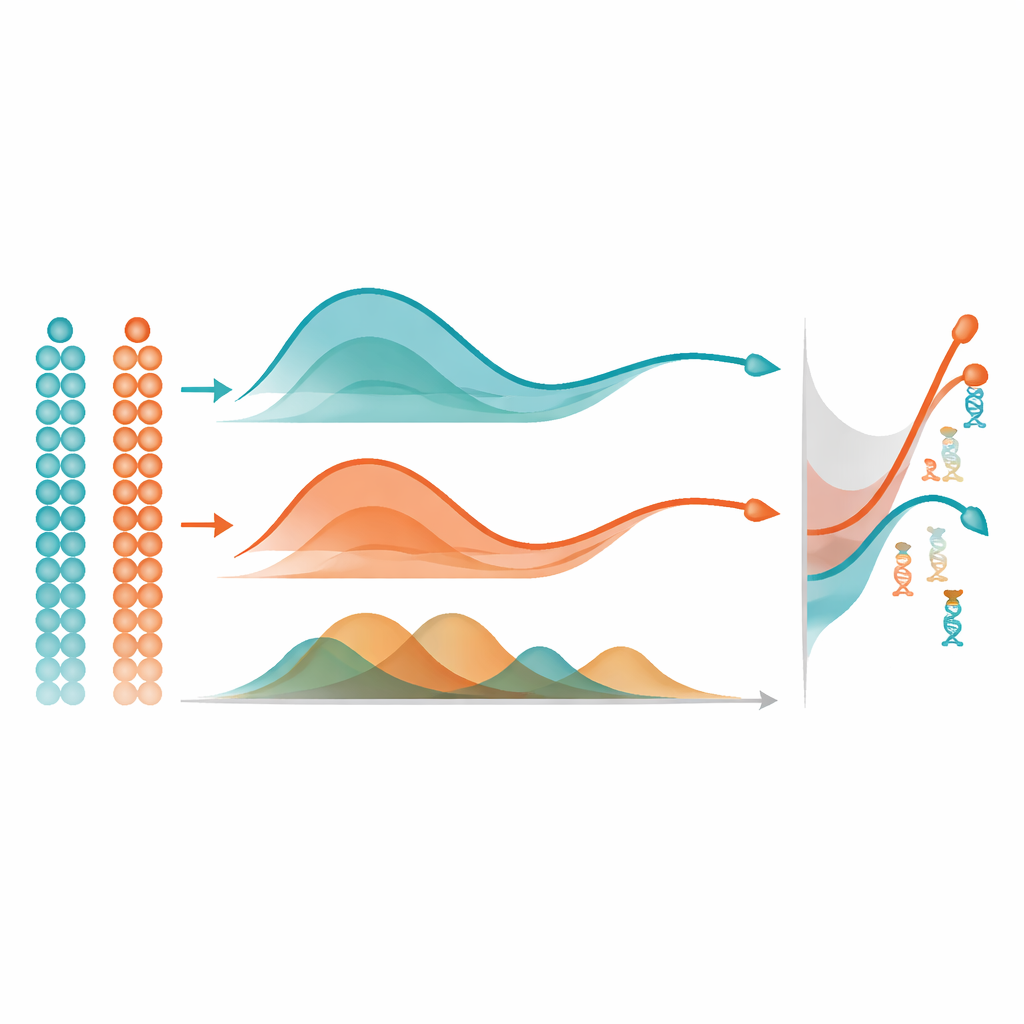
Una volta che mist ha appreso una traiettoria di metilazione liscia per ogni gene, usa un’idea semplice ma potente per trovare cambiamenti importanti: misura l’area tra le curve. In un singolo gruppo di cellule, confronta la traiettoria di un gene con una linea piatta per segnalare quelli che cambiano fortemente nel corso del pseudotempo. Nel confronto tra due gruppi, come due regioni cerebrali, allinea le due traiettorie e misura quanto siano distanti complessivamente, concentrandosi sulle differenze di forma piuttosto che solo sul livello basale. In ampie simulazioni numeriche, mist ha recuperato più accuratamente i veri schemi di metilazione sottostanti e ha identificato geni differenzialmente metilati meglio di alternative largamente usate come modelli additivi generalizzati e la regressione polinomiale standard. Ha anche superato un altro metodo di metilazione che ignora il pseudotempo, evidenziando il valore di modellare esplicitamente l’ordinamento temporale delle cellule.
Vedere lo sviluppo attraverso gli occhi epigenetici
Gli autori hanno applicato mist a dataset multi-omici reali per mostrare cosa significano questi guadagni statistici in termini biologici. Nell’embrione di topo, mist ha scoperto geni i cui schemi di metilazione seguivano transizioni di linea note, inclusi regolatori dello sviluppo cardiaco, della formazione delle cellule immunitarie e della perdita del potenziale delle cellule staminali. Nel tessuto cerebrale umano in sviluppo, ha rivelato geni la cui metilazione si è evoluta in modo diverso nella corteccia frontale e nell’ippocampo attraverso stadi dalla gestazione all’età adulta. Per esempio, un gene centrale per la segnalazione legata alla memoria mostrava una diminuzione della metilazione lungo la traiettoria ippocampale, coerente con un aumento dell’attività in quest’area, mentre un recettore legato alla crescita nella corteccia frontale diventava più metilato con la maturazione del tessuto, suggerendo uno spostamento dalla crescita alla funzione a lungo termine. Questi risultati illustrano come mist possa collegare cambiamenti chimici sottili sul DNA a grandi variazioni nell’identità cellulare e nella circuiteria cerebrale.
Perché questo conta per la ricerca futura
Offrendo un modo fondato per seguire le dinamiche della metilazione del DNA nelle singole cellule nel tempo, mist colma un vuoto chiave nella cassetta degli attrezzi per lo studio dell’epigenoma. È progettato specificamente per dati di metilazione proporzionali, scarsi, e può evidenziare geni il cui comportamento regolatorio cambia mentre le cellule si sviluppano o divergono in diverse linee o regioni tissutali. Sebbene il metodo dipenda da buone stime del pseudotempo e possa essere intensivo in termini computazionali per regioni genomiche molto fini, è già praticabile a livello di gene o promotore ed è disponibile come software open-source. Per i non specialisti, il messaggio principale è che mist aiuta a tradurre enormi e rumorosi dataset di metilazione single-cell in mappe chiare di quando e dove si attivano interruttori regolatori importanti durante lo sviluppo, l’invecchiamento e la malattia.
Citazione: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Parole chiave: metilazione del DNA single-cell, analisi del pseudotempo, modellizzazione bayesiana, regolazione epigenetica, traiettorie dello sviluppo