Clear Sky Science · pl
mist: hierarchiczne bayesowskie ramy do wykrywania różnicowej dynamiki metylacji DNA w danych pojedynczych komórek
Śledząc znaki, które kształtują nasze komórki
Każda komórka w twoim ciele zawiera tę samą sekwencję DNA, a jednak komórki mózgowe, sercowe i odpornościowe zachowują się bardzo różnie. Jednym z powodów są chemiczne znaczniki na DNA, takie jak grupy metylowe, które pomagają włączać lub wyłączać geny. Dzięki nowym narzędziom naukowcy mogą teraz odczytywać te znaczniki w tysiącach pojedynczych komórek podczas ich rozwoju lub zmian. Ten artykuł przedstawia „mist” — metodę statystyczną, która przekształca te ogromne, zaszumione pomiary w czytelne opowieści o tym, jak te znaczniki DNA przesuwają się w czasie w rozwoju i chorobie.
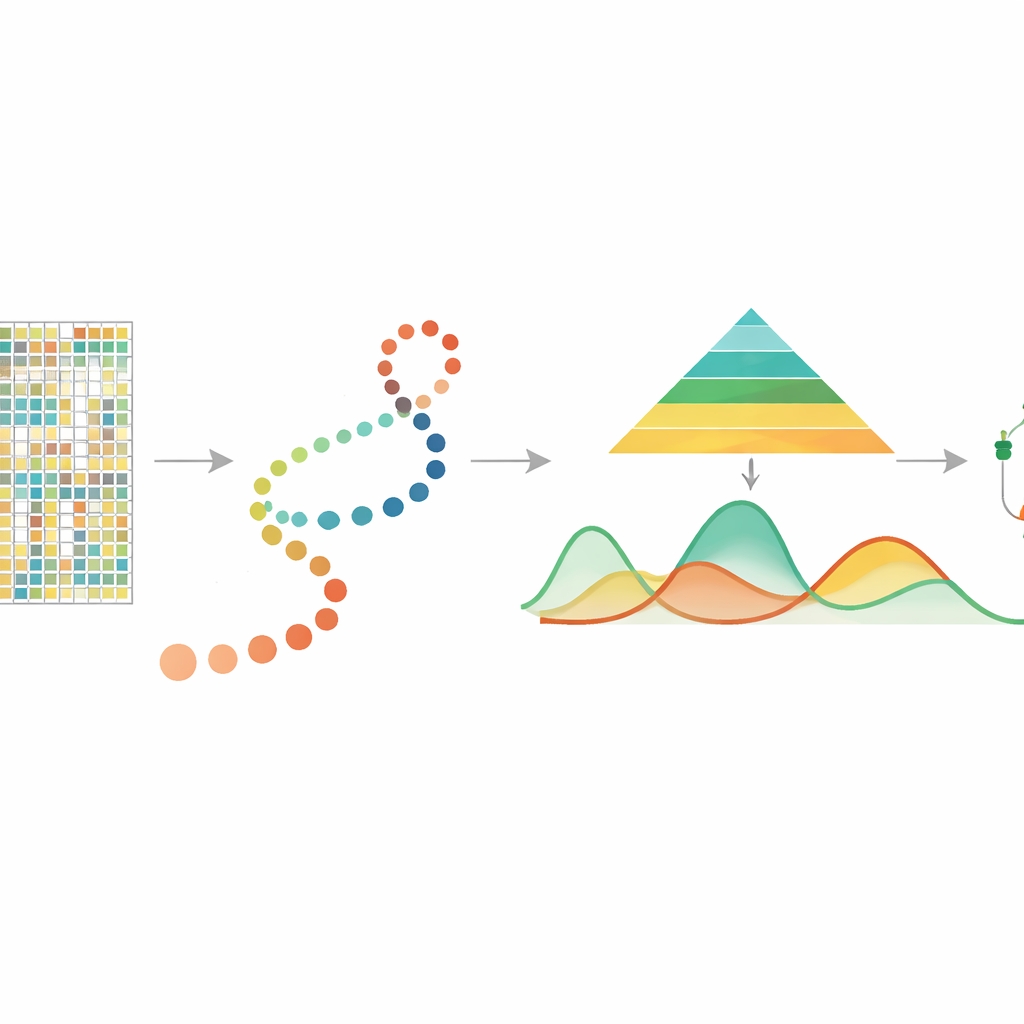
Odczytywanie chemicznych wskazówek na DNA
Metylacja DNA to małe chemiczne dołączenie do zasady cytozyny, często w miejscach nazywanych CpG, i odgrywa centralną rolę w kontrolowaniu, które geny są aktywne. Wcześniejsze badania na próbkach zbiorczych wykazały, że metylacja wiąże się ze starzeniem się, reakcjami na stres i rakiem oraz że może być dziedziczona przy podziałach komórkowych. Ostatnio technologie metylacji DNA pojedynczych komórek umożliwiły pomiar metylacji w każdej indywidualnej komórce, ujawniając bogate różnice między komórkami, które w analizie zbiorczej uległyby uśrednieniu. Jednak te pomiary są skąpe i zaszumione, i dotąd brakowało dedykowanego narzędzia do śledzenia, jak metylacja zmienia się ciągle w miarę postępu komórek w „pseudoczasie” — wywnioskowanej osi czasu, która porządkuje komórki od stanów wcześniejszych do późniejszych.
Podążanie za komórkami w niewidzialnej osi czasu
W wielu współczesnych eksperymentach badacze używają innych metod do oszacowania pseudoczasu, układając pojedyncze komórki wzdłuż ścieżki reprezentującej proces rozwojowy lub chorobowy. mist wykorzystuje to uporządkowanie komórek wraz z danymi o metylacji DNA z pojedynczych komórek pogrupowanymi według genów lub regionów, takich jak promotory, jako punkt wyjścia. Następnie modeluje poziom metylacji każdego genu jako gładką krzywą w funkcji pseudoczasu, pozwalając, by wielkość zmienności biologicznej różniła się między etapami wczesnymi i późnymi. To ważne, ponieważ wczesne etapy rozwojowe często są bardziej zróżnicowane i elastyczne, podczas gdy etapy późne są bardziej stabilne. Wbudowując te cechy w hierarchiczne ramy bayesowskie, mist potrafi oddzielić prawdziwe wzorce biologiczne od losowego szumu w bardzo skąpych danych.
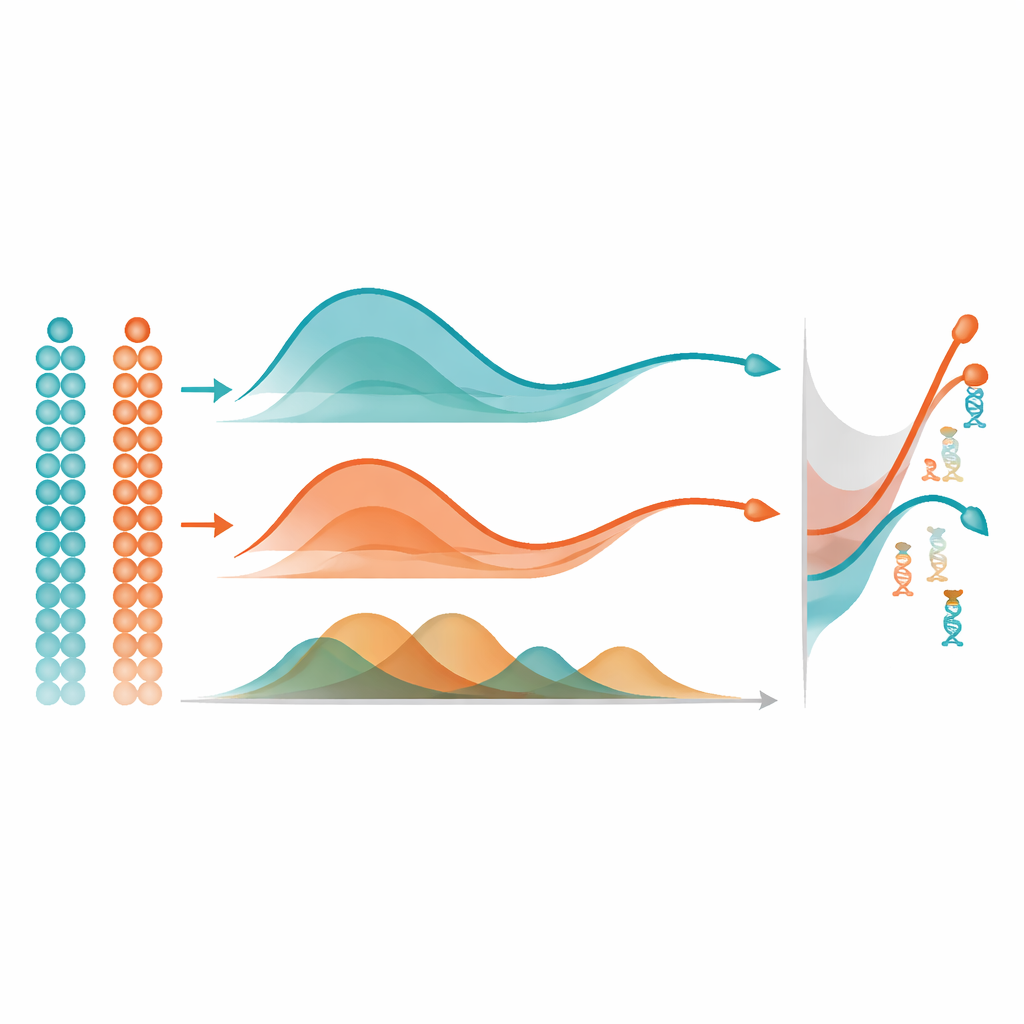
Z krzywych do kluczowych graczy genetycznych
Gdy mist nauczy się gładkiej trajektorii metylacji dla każdego genu, stosuje prosty, lecz silny pomysł, aby znaleźć istotne zmiany: mierzy pole między krzywymi. W pojedynczej grupie komórek porównuje trajektorię genu z linią stałą, by wskazać te, które silnie zmieniają się w pseudoczasie. Przy porównaniu dwóch grup, na przykład dwóch regionów mózgu, wyrównuje dwie trajektorie i mierzy, jak daleko są od siebie w całości, koncentrując się na różnicach w kształcie, a nie tylko w poziomie bazowym. W obszernej symulacji komputerowej mist dokładniej odtwarzał prawdziwe ukryte wzorce metylacji i identyfikował geny różnicowo metylowane niż powszechnie stosowane alternatywy, takie jak uogólnione modele addytywne i standardowa regresja wielomianowa. Przewyższał też inną metodę metylacji, która ignoruje pseudoczas, podkreślając wartość jawnego modelowania porządku czasowego komórek.
Widzieć rozwój przez epigenetyczne oko
Autorzy zastosowali mist do rzeczywistych zestawów danych multi-omics, aby pokazać, co te statystyczne korzyści oznaczają biologicznie. W embrionach myszy mist odkrył geny, których wzorce metylacji śledziły znane przejścia linii komórkowych, w tym regulatorów rozwoju serca, powstawania komórek odpornościowych oraz utraty potencjału komórek macierzystych. W rozwijającej się tkance mózgowej człowieka ujawnił geny, których metylacja ewoluowała inaczej w korze przedczołowej i hipokampie na etapach od okresu prenatalnego do dorosłości. Na przykład gen kluczowy dla sygnalizacji związanej z pamięcią wykazywał malejącą metylację wzdłuż trajektorii hipokampa, co odpowiadałoby rosnącej aktywności w tym regionie, podczas gdy receptor związany z wzrostem w korze przedczołowej stał się bardziej metylowany w miarę dojrzewania tkanki, sugerując przesunięcie od wzrostu w kierunku funkcji długoterminowych. Te odkrycia ilustrują, jak mist może łączyć subtelne zmiany chemiczne na DNA z dużymi zmianami tożsamości komórek i obwodów mózgowych.
Dlaczego to ma znaczenie dla przyszłych badań
Oferując zasadniczy sposób śledzenia dynamiki metylacji DNA w pojedynczych komórkach w czasie, mist wypełnia istotną lukę w zestawie narzędzi do badania epigenomu. Został zaprojektowany specjalnie dla skąpych, proporcjonalnych danych metylacyjnych i potrafi wyróżnić geny, których zachowanie regulatorowe zmienia się, gdy komórki rozwijają się lub rozdzielają na różne linie lub regiony tkankowe. Chociaż metoda zależy od dobrych oszacowań pseudoczasu i może być obliczeniowo intensywna dla bardzo drobnych regionów genomowych, jest już praktyczna na poziomie genu lub promotora i dostępna jako oprogramowanie open-source. Dla osób niebędących specjalistami główne przesłanie jest takie, że mist pomaga przetłumaczyć ogromne, zaszumione zbiory danych metylacji pojedynczych komórek na czytelne mapy, kiedy i gdzie przełączane są ważne regulatory podczas rozwoju, starzenia i choroby.
Cytowanie: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Słowa kluczowe: metylacja DNA pojedynczych komórek, analiza pseudoczasu, modelowanie bayesowskie, regulacja epigenetyczna, trajektorie rozwojowe