Clear Sky Science · ru
mist: иерархическая байесовская система для обнаружения различий в динамике метилирования ДНК в одно-клеточных данных
Следуя меткам, формирующим наши клетки
Каждая клетка в вашем организме содержит одинаковую ДНК, однако клетки мозга, сердца и иммунной системы ведут себя очень по-разному. Одна из причин — химические метки на ДНК, например метильные группы, которые помогают включать или выключать гены. С новыми технологиями учёные теперь могут считывать эти метки в тысячах отдельных клеток по мере их развития или изменения. В этой статье представлена «mist» — статистический метод, который превращает эти огромные, зашумлённые измерения в понятные истории о том, как эти метки на ДНК меняются во времени при развитии и в болезни.
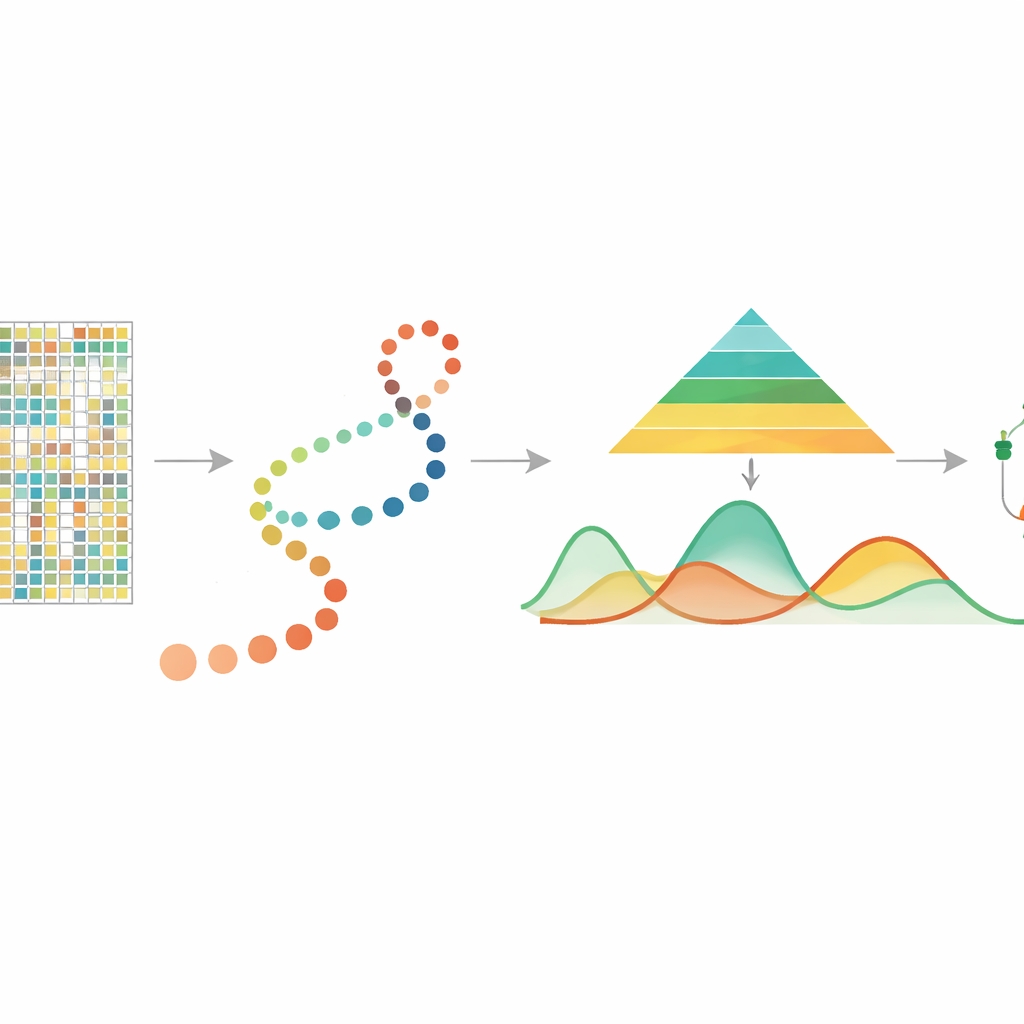
Чтение химических подсказок на ДНК
Метилирование ДНК — это небольшое химическое присоединение к букве цитозин, часто в сайтах, называемых CpG, и оно играет ключевую роль в контроле активности генов. Предыдущие исследования на массовых образцах показали, что метилирование связано со старением, реакциями на стресс и раком и что оно может наследоваться при делении клеток. В последнее время технологии одно-клеточного метилирования ДНК позволили измерять метилирование в каждой отдельной клетке, выявляя богатое разнообразие между клетками, которое в массовых образцах усреднилось бы. Однако эти измерения разрежены и шумны, и до сих пор не было специализированного инструмента, позволяющего проследить, как метилирование меняется непрерывно по мере продвижения клеток вдоль «псевдовремени» — выведённой шкалы, упорядочивающей клетки от более ранних к более поздним состояниям.
Следование клеткам по невидимой временной линии
Во многих современных экспериментах исследователи используют другие методы для оценки псевдовремени, располагая отдельные клетки вдоль пути, который отражает процесс развития или заболевания. mist берет это упорядочение клеток вместе с одно-клеточными данными по метилированию ДНК, сгруппированными по генам или регионам, таким как промоторы, в качестве исходных данных. Затем метод моделирует уровень метилирования каждого гена как гладкую кривую по псевдовремени, позволяя величине биологической вариабельности различаться между ранними и поздними стадиями. Это важно, потому что ранние стадии развития часто более разнообразны и гибки, тогда как поздние стадии более стабильны. Встраивая эти особенности в иерархическую байесовскую модель, mist может отделять истинные биологические паттерны от случайного шума в сильно разреженных данных.
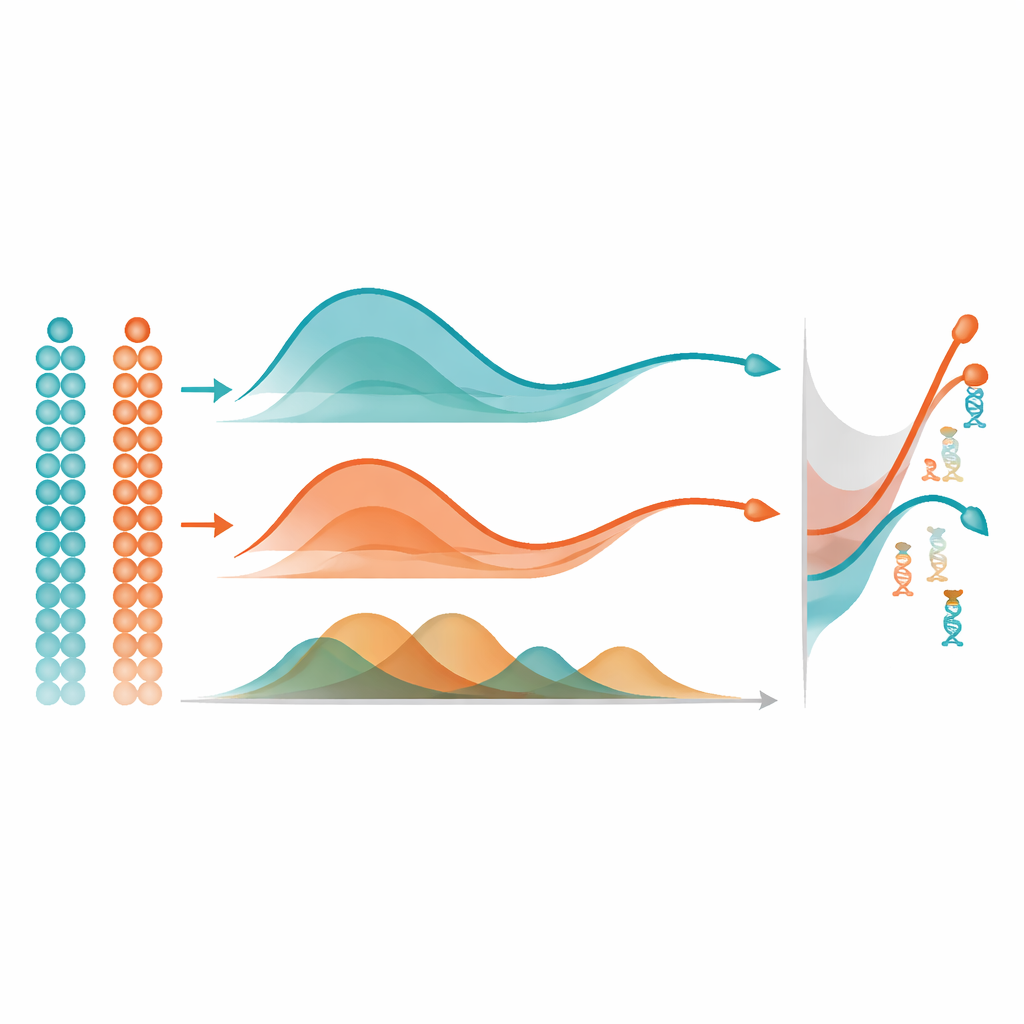
От кривых к ключевым генам
Когда mist выучивает гладкую траекторию метилирования для каждого гена, он использует простую, но мощную идею для выявления важных изменений: измеряет площадь между кривыми. В одной группе клеток он сравнивает траекторию гена с плоской линией, чтобы пометить те, которые существенно меняются по псевдовремени. При сравнении двух групп, например двух областей мозга, он выравнивает обе траектории и измеряет, насколько они в целом удалены друг от друга, сосредотачиваясь на различиях в форме, а не только в базовом уровне. В обширных компьютерных симуляциях mist точнее восстанавливал истинные подлежащие паттерны метилирования и выявлял дифференциально метилированные гены по сравнению с широко используемыми альтернативами, такими как обобщённые аддитивные модели и стандартная полиномиальная регрессия. Он также превосходил другой метод для анализа метилирования, игнорирующий псевдовремя, подчёркивая ценность явного моделирования временного порядка клеток.
Видение развития сквозь эпигенетические призмы
Авторы применили mist к реальным мультиомным наборам данных, чтобы показать, что означают эти статистические преимущества с биологической точки зрения. В эмбрионах мыши mist выявил гены, чьи паттерны метилирования соответствовали известным переходам между линиями клеток, включая регуляторы развития сердца, формирования иммунных клеток и утраты потенциала стволовых клеток. В развивающейся ткани человеческого мозга он обнаружил гены, метилирование которых развивалось по-разному в лобной коре и гиппокампе на этапах от внутриутробного развития до взрослости. Например, ген, центральный для сигнальных путей, связанных с памятью, показал снижение метилирования вдоль гиппокампальной траектории, что согласуется с усилением активности в этой области, тогда как рецептор, связанный с ростом, в лобной коре стал более метилированным по мере созревания ткани, что указывает на переход от роста к длительной функции. Эти находки иллюстрируют, как mist может связывать тонкие химические изменения на ДНК с крупными сдвигами в идентичности клеток и мозговой схемотехнике.
Почему это важно для будущих исследований
Предоставляя принципиальный способ отслеживать динамику метилирования ДНК в отдельных клетках с течением времени, mist заполняет ключевой пробел в инструментарии для изучения эпигенома. Он специально разработан для разреженных пропорциональных данных по метилированию и может выделять гены, регуляторное поведение которых меняется по мере развития клеток или их расхождения в разные линии или тканевые области. Хотя метод зависит от достоверных оценок псевдовремени и может быть вычислительно затратным для очень тонких геномных регионов, он уже практичен на уровне генов или промоторов и доступен как программное обеспечение с открытым исходным кодом. Для неспециалистов главный вывод в том, что mist помогает превратить огромные, зашумлённые наборы данных по одно-клеточному метилированию в понятные карты того, когда и где во время развития, старения и болезни переключаются важные регуляторные механизмы.
Цитирование: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Ключевые слова: метилирование ДНК в отдельных клетках, анализ псевдовремени, байесовское моделирование, эпигенетическая регуляция, развитийные траектории