Clear Sky Science · sv
mist: ett hierarkiskt Bayesianskt ramverk för att upptäcka differential DNA-metyleringsdynamik i enkelcellsdata
Följa märkena som formar våra celler
Varje cell i din kropp bär på samma DNA, ändå beter sig nervceller, hjärtceller och immunceller mycket olika. En förklaring är kemiska märken på DNA, såsom metyleringsgrupper, som hjälper till att slå av och på gener. Med nya verktyg kan forskare nu läsa dessa märken i tusentals enskilda celler medan de utvecklas eller förändras. Denna artikel presenterar ”mist”, en statistisk metod som förvandlar dessa massiva, brusiga mätningar till tydliga berättelser om hur dessa DNA-märken skiftar över tid vid utveckling och sjukdom.
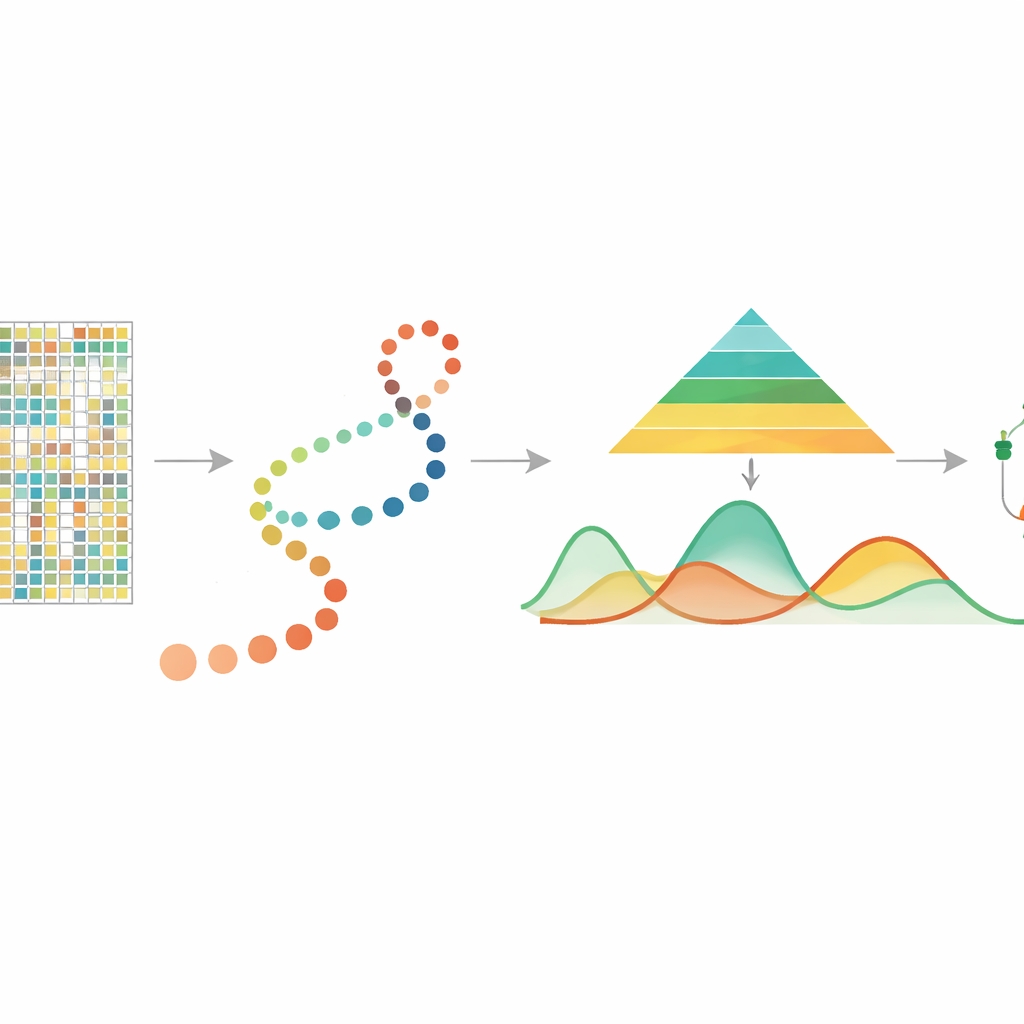
Avkoda kemiska spår på DNA
DNA-metylering är en liten kemisk tillsats på DNA-basen cytosin, ofta vid platser som kallas CpG, och spelar en central roll i att kontrollera vilka gener som är aktiva. Tidigare studier med bulkprov visade att metylering är involverad i åldrande, stressreaktioner och cancer, och att den kan ärvas när celler delar sig. På senare tid har tekniker för enkelcells-DNA-metylering gjort det möjligt att mäta metylering i varje enskild cell, vilket avslöjar rika skillnader mellan celler som annars skulle jämnas ut i bulk. Dessa mätningar är dock glesa och brusiga, och fram till nu har det saknats ett särskilt verktyg för att följa hur metylering förändras kontinuerligt när celler rör sig genom ett utvecklings-"pseudotid" — en härledd tidslinje som ordnar celler från tidigare till senare tillstånd.
Följa celler längs en osynlig tidslinje
I många moderna experiment använder forskare andra metoder för att uppskatta pseudotid och ordnar enskilda celler längs en bana som representerar en utvecklings- eller sjukdomsprocess. mist tar denna cellordning, tillsammans med enkelcells-DNA-metyleringsdata grupperade efter gener eller regioner som promotorer, som utgångspunkt. Den modellerar sedan metyleringsnivån för varje gen som en slät kurva över pseudotid och tillåter att mängden biologisk variabilitet skiljer sig mellan tidiga och sena stadier. Detta är viktigt eftersom tidiga utvecklingsstadier ofta är mer varierade och flexibla, medan senare stadier är mer stabila. Genom att bygga in dessa egenskaper i ett hierarkiskt Bayesianskt ramverk kan mist skilja verkliga biologiska mönster från slumpmässigt brus i mycket glesa data.
Från kurvor till viktiga genetiska aktörer
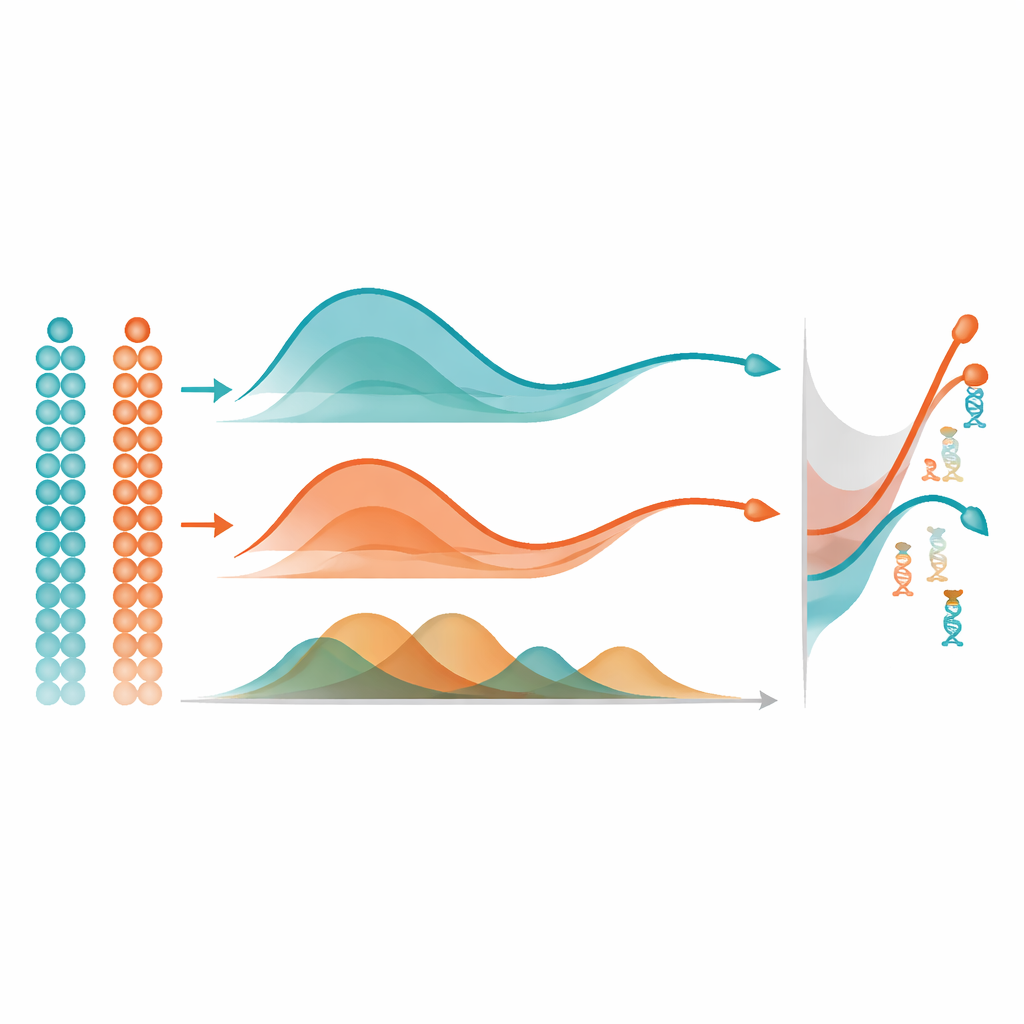
När mist lärt sig en slät metyleringstrajektoria för varje gen använder den en enkel men kraftfull idé för att hitta viktiga förändringar: den mäter arean mellan kurvor. I en enskild cellgrupp jämför den en gens trajectory med en horisontell linje för att flagga gener som förändras kraftigt över pseudotid. Vid jämförelse av två grupper, såsom två hjärnregioner, justerar den de två trajektorierna och mäter hur långt ifrån varandra de ligger totalt, med fokus på skillnader i form snarare än enbart i baslinjenivå. I omfattande computersimuleringar återvann mist mer exakt de verkliga underliggande metyleringsmönstren och identifierade differentierat metylerade gener bättre än ofta använda alternativ som generaliserade additiva modeller och standard polynomregression. Den överträffade också en annan metyleringsmetod som ignorerar pseudotid, vilket understryker värdet av att uttryckligen modellera cellernas temporala ordning.
Se utveckling genom epigenetiska ögon
Författarna tillämpade mist på verkliga multi-omiska datasätt för att visa vad dessa statistiska förbättringar betyder biologiskt. I musembryon upptäckte mist gener vars metyleringsmönster följde kända linjeskiften, inklusive regulatorer för hjärtutveckling, bildandet av immunceller och förlusten av stamcells potential. I utvecklande mänsklig hjärnvävnad visade den gener vars metylering utvecklades olika i frontalloben och hippocampus över stadier från gestation till vuxen ålder. Till exempel visade en gen central för minnesrelaterad signalering avtagande metylering längs hippocampal trajektoria, förenligt med ökande aktivitet i denna region, medan en tillväxtrelaterad receptor i frontalloben blev mer metylerad när vävnaden mognade, vilket antyder en förskjutning från tillväxt till långsiktig funktion. Dessa fynd illustrerar hur mist kan koppla subtila kemiska förändringar på DNA till stora skiften i cellidentitet och hjärnkretsar.
Varför detta är viktigt för framtida forskning
Genom att erbjuda ett principfast sätt att följa DNA-metyleringsdynamik i enskilda celler över tid fyller mist en viktig lucka i verktygslådan för att studera epigenomet. Den är speciellt utformad för gles, proportionell metyleringsdata och kan lyfta fram gener vars regulatoriska beteende förändras när celler utvecklas eller divergerar in i olika linjer eller vävnadsregioner. Även om metoden är beroende av bra pseudotidsuppskattningar och kan vara beräkningsmässigt krävande för mycket fina genomiska regioner, är den redan praktisk på gen- eller promotor-nivå och finns tillgänglig som öppen källkod. För icke-specialister är huvudbudskapet att mist hjälper till att översätta enorma, brusiga enkelcells-metyleringsdatasätt till tydliga kartor över när och var viktiga regulatoriska strömbrytare slås på eller av under utveckling, åldrande och sjukdom.
Citering: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Nyckelord: single-cell DNA methylation, pseudotime analysis, Bayesian modeling, epigenetic regulation, developmental trajectories