Clear Sky Science · en
mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data
Following the Marks That Shape Our Cells
Every cell in your body carries the same DNA, yet brain cells, heart cells, and immune cells behave very differently. One reason is chemical tags on DNA, such as methyl groups, that help switch genes on or off. With new tools, scientists can now read these tags in thousands of individual cells as they develop or change. This article introduces “mist,” a statistical method that turns those massive, noisy measurements into clear stories about how these DNA tags shift over time in development and disease.
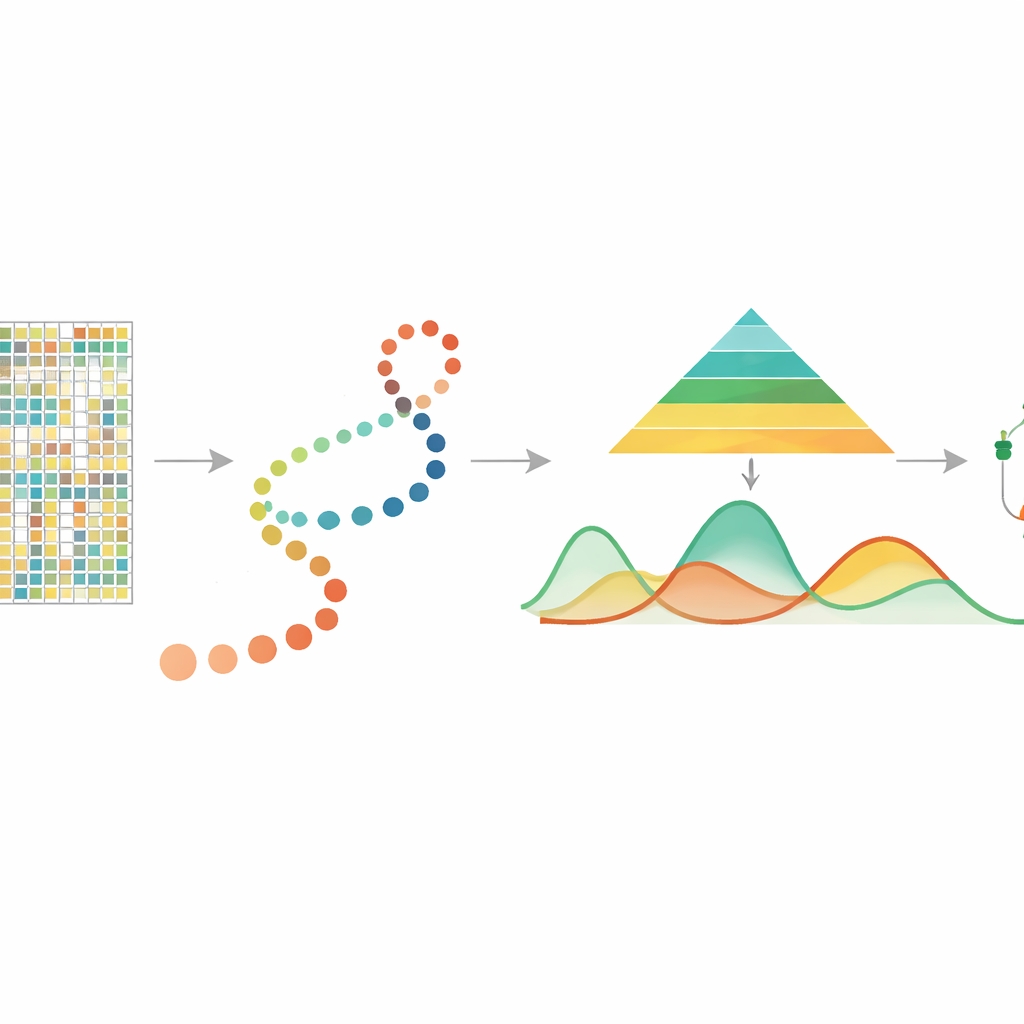
Reading Chemical Clues on DNA
DNA methylation is a small chemical addition to the DNA letter cytosine, often at sites called CpGs, and it plays a central role in controlling which genes are active. Past studies using bulk samples showed that methylation is involved in aging, stress responses, and cancer, and that it can be inherited when cells divide. Recently, single-cell DNA methylation technologies have made it possible to measure methylation for each individual cell, revealing rich differences among cells that would be averaged away in bulk. However, these measurements are sparse and noisy, and until now there has been no dedicated tool to follow how methylation changes continuously as cells progress through developmental “pseudotime” — an inferred timeline that orders cells from earlier to later states.
Following Cells Along an Invisible Timeline
In many modern experiments, researchers use other methods to estimate pseudotime, arranging individual cells along a path that represents a developmental or disease process. mist takes this cell ordering, together with single-cell DNA methylation data grouped by genes or regions such as promoters, as its starting point. It then models the methylation level of each gene as a smooth curve over pseudotime, allowing the amount of biological variability to differ between early and late stages. This is important because early developmental stages are often more diverse and flexible, whereas later stages are more stable. By building these features into a hierarchical Bayesian framework, mist can separate true biological patterns from random noise in highly sparse data.
From Curves to Key Genetic Players
Once mist has learned a smooth methylation trajectory for each gene, it uses a simple yet powerful idea to find important changes: it measures the area between curves. In a single group of cells, it compares a gene’s trajectory to a flat line to flag those that change strongly over pseudotime. When comparing two groups, such as two brain regions, it aligns the two trajectories and measures how far apart they are overall, focusing on differences in shape rather than just baseline level. In extensive computer simulations, mist more accurately recovered the true underlying methylation patterns and identified differentially methylated genes than widely used alternatives like generalized additive models and standard polynomial regression. It also outperformed another methylation method that ignores pseudotime, highlighting the value of explicitly modeling the temporal ordering of cells.
Seeing Development Through Epigenetic Eyes
The authors applied mist to real multi-omics datasets to show what these statistical gains mean biologically. In mouse embryos, mist uncovered genes whose methylation patterns tracked known lineage transitions, including regulators of heart development, immune cell formation, and the loss of stem cell potential. In developing human brain tissue, it revealed genes whose methylation evolved differently in the frontal cortex and hippocampus across stages from gestation to adulthood. For example, a gene central to memory-related signaling showed decreasing methylation along the hippocampal trajectory, consistent with increasing activity in this region, while a growth-related receptor in the frontal cortex became more methylated as the tissue matured, suggesting a shift from growth to long-term function. These findings illustrate how mist can connect subtle chemical changes on DNA to major shifts in cell identity and brain circuitry.
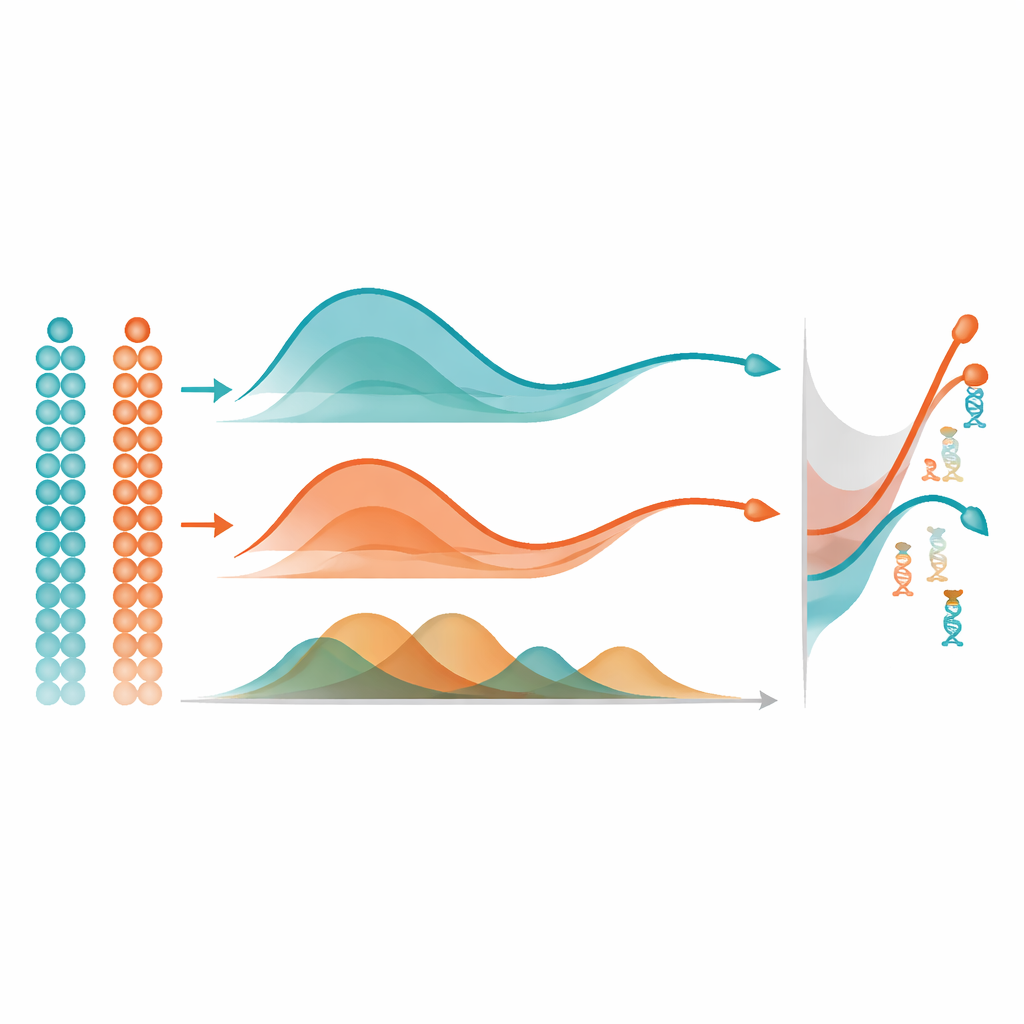
Why This Matters for Future Research
By offering a principled way to follow DNA methylation dynamics in single cells over time, mist fills a key gap in the toolkit for studying the epigenome. It is designed specifically for sparse, proportional methylation data and can highlight genes whose regulatory behavior changes as cells develop or diverge into different lineages or tissue regions. Although the method depends on good pseudotime estimates and can be computationally intensive for very fine genomic regions, it is already practical at the gene or promoter level and is available as open-source software. For non-specialists, the main message is that mist helps translate enormous, noisy single-cell methylation datasets into clear maps of when and where important regulatory switches are flipped during development, aging, and disease.
Citation: Duan, D., Ma, W., Tang, W. et al. mist: a hierarchical Bayesian framework for detecting differential DNA methylation dynamics in single-cell data. Nat Commun 17, 3835 (2026). https://doi.org/10.1038/s41467-026-70523-y
Keywords: single-cell DNA methylation, pseudotime analysis, Bayesian modeling, epigenetic regulation, developmental trajectories