Clear Sky Science · sv
En STDFT-CEEMD-metod med vågpakettröskling för exonprediktion i eukaryota celler
Att hitta de användbara delarna av vår genetiska kod
Inuti varje cell bär långa DNA-strängar instruktioner för att bygga de proteiner som håller oss vid liv. Men endast vissa avsnitt av detta DNA kodar faktiskt för proteiner, medan stora delar fungerar mer som interpunktion eller bakgrund. Denna artikel tar sig an en central utmaning i modern genetik: hur man på ett tillförlitligt sätt lokaliserar de protein-kodande delarna, kallade exoner, i enorma mängder råa DNA-data med hjälp av smarta signalbehandlingsverktyg hämtade från ingenjörsvetenskapen.
Varför det är viktigt att skilja signal från brus
Gener hos människor och andra komplexa organismer är uppdelade i exoner, som bär användbar information, och introner, som inte gör det. Under proteinproduktion kopierar cellerna DNA till RNA och klipper sedan bort intronerna, för att sy ihop exoner till ett slutligt budskap som bestämmer ett proteins sammansättning. Att identifiera var exoner börjar och slutar är avgörande för att förstå hur gener fungerar, hur sjukdomar uppstår och hur behandlingar kan anpassas. Traditionella datorbaserade metoder förlitar sig ofta tungt på stora, noggrant annoterade träningsdata eller detaljerade biologiska modeller, som inte alltid finns eller kan misslyckas för dåligt studerade arter. Därför är metoder som kan arbeta direkt på rått DNA, och betrakta det som en signal att analysera, allt mer attraktiva.
Att förvandla DNA till en signal
I denna studie behandlar författarna DNA som om det vore en vågform, lik en ljudspåra, och tillämpar sedan en rad bearbetningssteg. Först omvandlas varje av de fyra DNA-bokstäverna till siffror med ett särskilt schema baserat på Hadamard-matriser, som är noggrant utvalda mönster av +1 och −1. Detta steg skapar fyra rena numeriska spår som bevarar all information från den ursprungliga sekvensen men som är bättre lämpade för analys. Därefter skannar metoden längs sekvensen med ett glidande fönster och använder ett tids–frekvensverktyg kallat Short-Time Discrete Fourier Transform för att söka efter ett återkommande mönster som uppträder var tredje bas. Denna “period-3”-rytm är ett välkänt kännetecken för protein-kodande regioner eftersom proteiner byggs av trebokstavsord, eller kodon, i den genetiska koden.
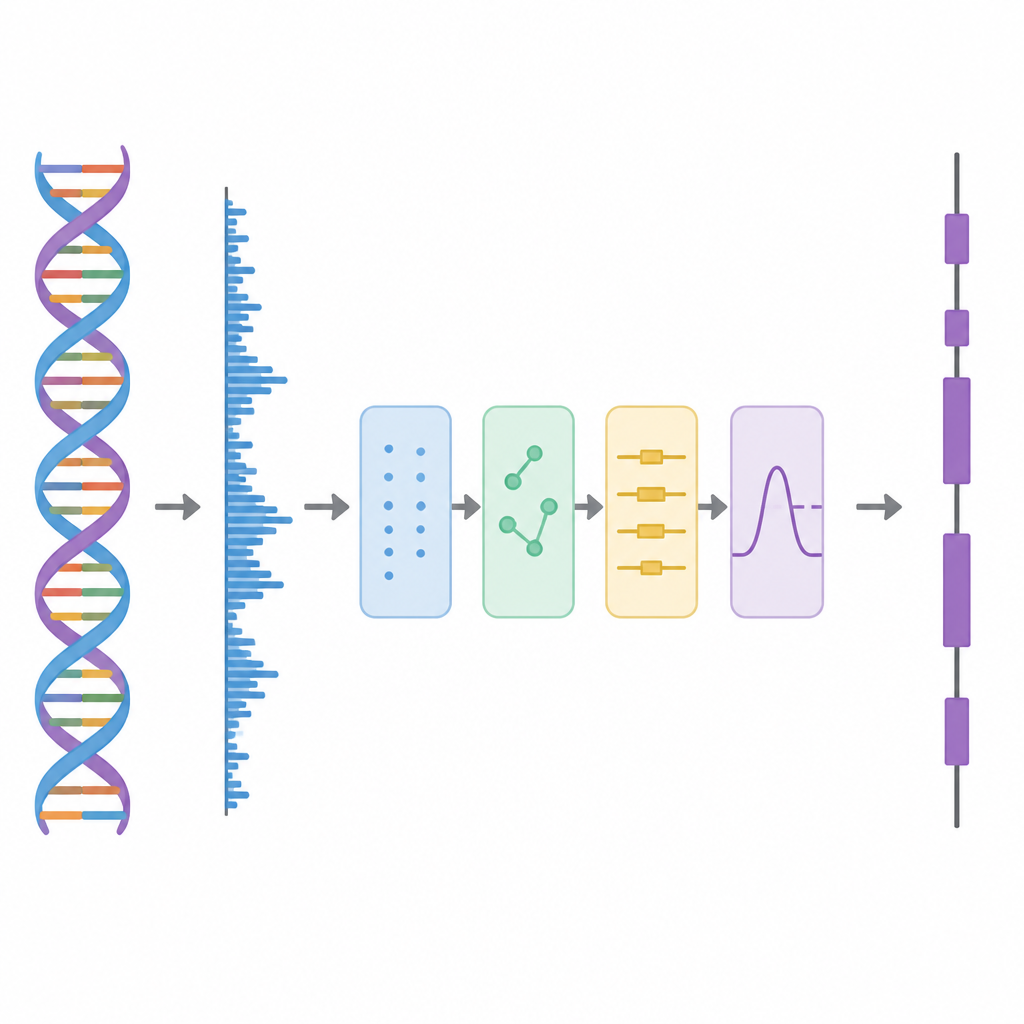
Att skala bort signalens lager
Verkliga genomdata är röriga. Långsiktiga bakgrundstrender och slumpmässiga fluktuationer kan sudda ut period-3-mönstret, särskilt för korta exoner. För att hantera detta använder författarna en idé från avancerad signaldekomposition, där en komplicerad vågform delas upp i enklare byggstenar. De använder en teknik som kallas Complete Ensemble Empirical Mode Decomposition, vilken upprepade gånger lägger till noggrant balanserat brus och sedan medelvärdesbildar resultaten för att framställa en uppsättning renare komponenter. Ett självkorrelationsmått används därefter för att avgöra vilka av dessa komponenter som bär meningsfull struktur och vilka som domineras av brus. De brusdominerade delarna renas vidare med hjälp av vågpakettröskling, en metod som tar bort små, ryckiga variationer samtidigt som den huvudsakliga signalformen bevaras.
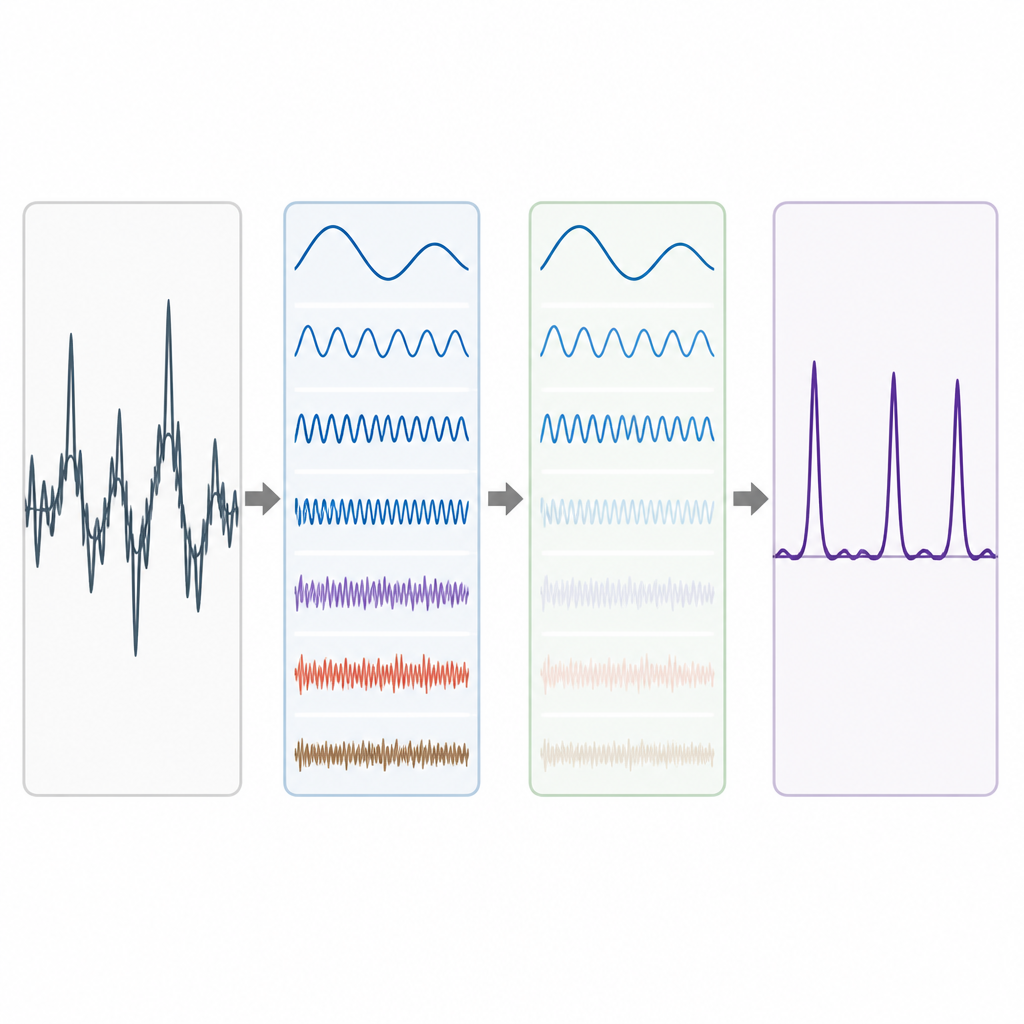
Test av metoden på verkliga gener
För att se hur väl deras pipeline fungerar tillämpar författarna den på väldokumenterade gener från rundmasken Caenorhabditis elegans och husmusen, samt på ett referensset bestående av 195 gensegment från människa, mus och råtta. I varje fall jämför de sina exonprediktioner med expertannoteringar. Deras angreppssätt ger tydligare toppar där verkliga exoner förekommer och lägre bakgrund i regioner som inte kodar för proteiner. När de sammanfattar prestanda med vanliga mått som känslighet, specificitet, noggrannhet och area under ROC-kurvan, överträffar deras metod konsekvent flera tidigare signalbehandlingsmetoder som förlitar sig på enklare filter eller mindre raffinerade dekompositioner. Vinsterna är särskilt märkbara i balansen mellan korrekt upptäckt av exoner och undvikande av falska larm.
Vad detta innebär för genomisk analys
Huvudbudskapet för läsaren är att författarna har byggt en mer precis ”lyssningsanordning” för genomet. Genom att noggrant mappa DNA till siffror, följa dess rytmer över korta fönster, skala isär signalen i rena komponenter och målmedvetet avlägsna brus, får de en mycket skarpare bild av var de protein-kodande instruktionerna ligger. Även om den nuvarande implementationen kan vara beräkningsmässigt krävande och fortfarande kräver inpassning av vissa parametrar, visar ramverket att verktyg från modern signalbehandling kan förbättra hur vi läser genomet i betydande grad. På längre sikt kan sådana metoder hjälpa forskare att annotera nya genomer snabbare och stödja efterföljande studier av genfunktion, sjukdomsmekanismer och personlig medicin.
Citering: Benarjee, S., Vaegae, N.K. A STDFT-CEEMD approach with wavelet packet thresholding for exon prediction in eukaryotic cells. Sci Rep 16, 15948 (2026). https://doi.org/10.1038/s41598-026-43722-2
Nyckelord: exonprediktion, genomisk signalbehandling, DNA-analys, protein-kodande regioner, brusreducering