Clear Sky Science · pl
Metoda STDFT-CEEMD z progowaniem pakietów falkowych do predykcji eksonów w komórkach eukariotycznych
Odnajdywanie użytecznych fragmentów naszego kodu genetycznego
W każdej komórce długie łańcuchy DNA niosą instrukcje budowy białek, które podtrzymują życie. Tylko jednak niektóre odcinki tego DNA rzeczywiście kodują białka, podczas gdy rozległe fragmenty pełnią rolę interpunkcji lub tła. Artykuł ten podejmuje kluczowe wyzwanie współczesnej genetyki: jak wiarygodnie wykrywać fragmenty kodujące białka, zwane eksonami, w ogromnych zasobach surowych danych DNA, używając inteligentnych narzędzi przetwarzania sygnałów zaczerpniętych z inżynierii.
Dlaczego rozdzielenie sygnału od szumu ma znaczenie
Geny u ludzi i innych złożonych organizmów są podzielone na eksony, które niosą użyteczne instrukcje, oraz introny, które tego nie robią. Podczas produkcji białka komórki przepisywują DNA na RNA, a następnie wycinają introny, łącząc eksony w końcową wiadomość determinującą budowę białka. Identyfikacja początku i końca eksonów jest kluczowa dla zrozumienia funkcji genów, mechanizmów chorób i możliwych terapii. Tradycyjne metody komputerowe często opierają się na dużych, starannie oznaczonych zbiorach treningowych albo na szczegółowych modelach biologicznych, które nie zawsze są dostępne lub zawodzą dla słabo zbadanych gatunków. Dlatego metody działające bezpośrednio na surowym DNA, traktując go jak sygnał do analizy, stają się coraz bardziej atrakcyjne.
Przekształcanie DNA w sygnał
W tym badaniu autorzy traktują DNA jak przebieg falowy, podobny do ścieżki dźwiękowej, i stosują po kolei kilka etapów przetwarzania. Najpierw każda z czterech liter DNA jest przekształcana w liczby przy użyciu schematu opartego na macierzach Hadamarda, czyli starannie dobranych wzorcach plusów i minusów. Ten etap tworzy cztery czyste ścieżki numeryczne, które zachowują wszystkie informacje z oryginalnej sekwencji, ale są lepiej przystosowane do analizy. Następnie metoda przesuwa okno wzdłuż sekwencji i używa narzędzia czas-częstotliwość o nazwie krótkoczasowa dyskretna transformata Fouriera (STDFT), by wyszukać powtarzający się wzorzec pojawiający się co trzy zasady. Ten rytm „okres-3” jest dobrze znaną cechą obszarów kodujących białka, ponieważ białka budowane są ze słów trójliterowych, czyli kodonów, w kodzie genetycznym.
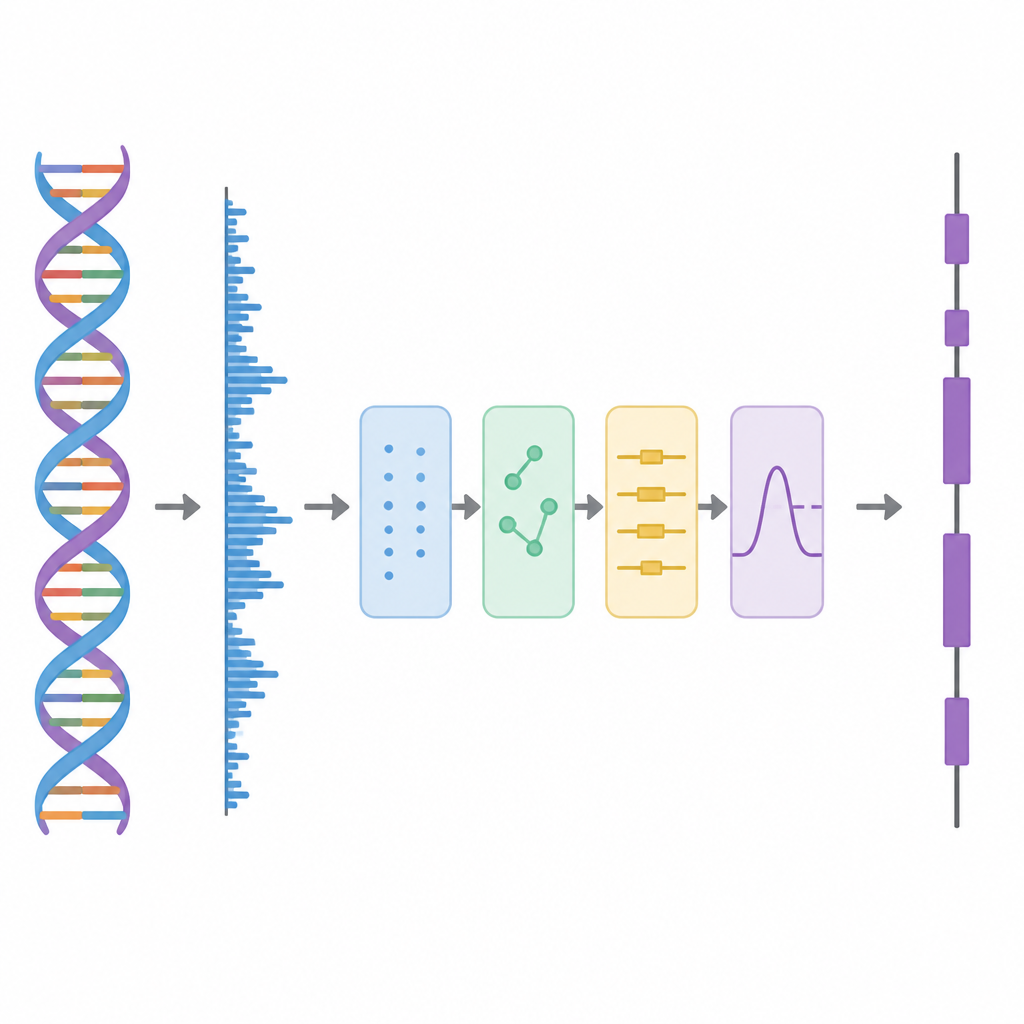
Oddzielanie warstw sygnału
Prawdziwe dane genomowe są chaotyczne. Długozasięgowe trendy tła i losowe fluktuacje mogą rozmazać wzorzec okres-3, szczególnie w przypadku krótkich eksonów. Aby temu sprostać, autorzy wykorzystują pomysł z zaawansowanego rozkładu sygnału, w którym skomplikowany przebieg jest dzielony na prostsze składniki. Stosują technikę zwaną Complete Ensemble Empirical Mode Decomposition (CEEMD), która wielokrotnie dodaje starannie zbalansowany szum, a potem uśrednia wyniki, by uzyskać zestaw czyściejszych komponentów. Miara autokorelacji jest następnie używana do decyzji, które z tych składników niosą istotną strukturę, a które są zdominowane przez szum. Zaszumione fragmenty są dodatkowo oczyszczane przy użyciu progowania w pakietach falkowych, metody usuwającej drobne, drgające zmiany przy zachowaniu głównego kształtu sygnału.
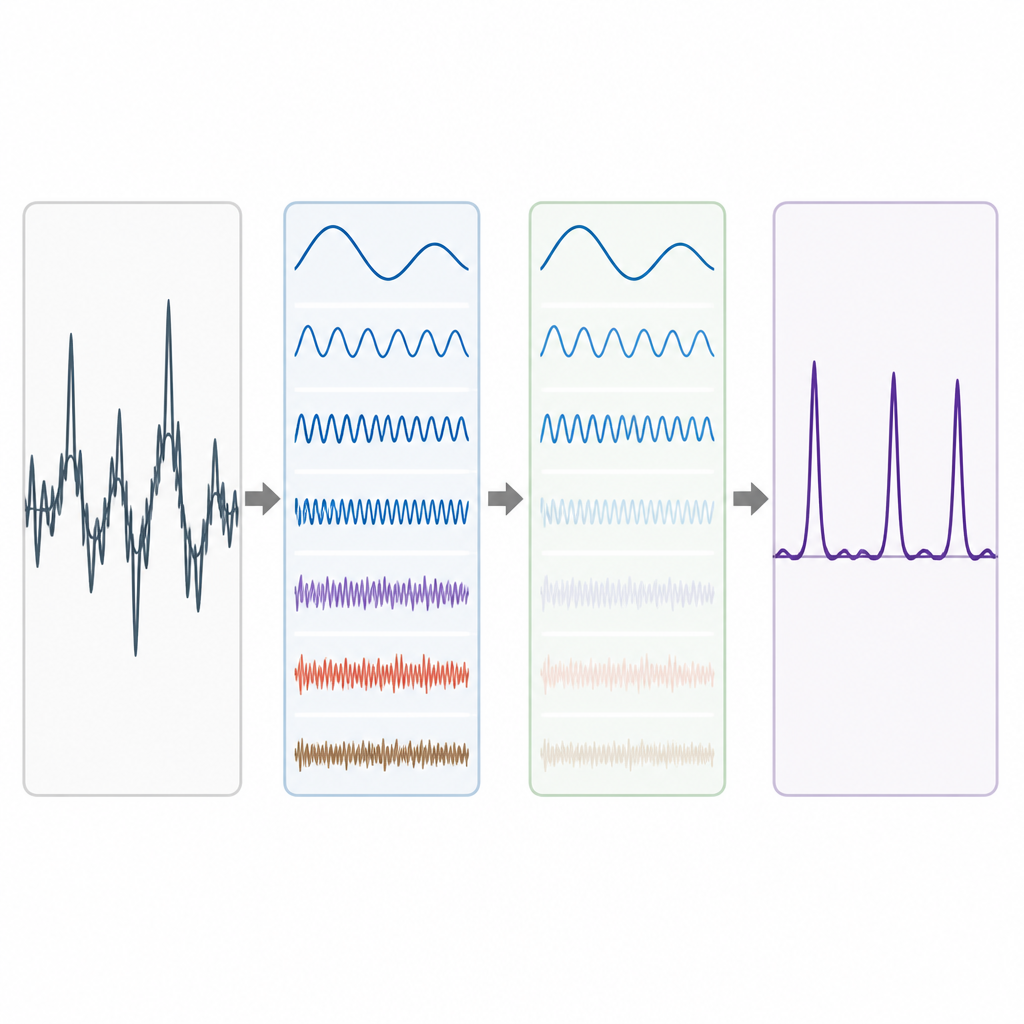
Testowanie metody na prawdziwych genach
Aby ocenić skuteczność całego procesu, autorzy zastosowali go do dobrze zbadanych genów nicienia Caenorhabditis elegans i myszy domowej, oraz do zestawu referencyjnego 195 fragmentów genów z człowieka, myszy i szczura. W każdym przypadku porównywali swoje predykcje eksonów z eksperckimi adnotacjami. Ich podejście daje czyściejsze szczyty w miejscach, gdzie występują prawdziwe eksony, oraz niższe tło w regionach niekodujących. Podsumowując wydajność za pomocą miar takich jak czułość, swoistość, dokładność i pole pod krzywą ROC, metoda konsekwentnie przewyższa kilka wcześniejszych podejść opartych na prostszych filtrach lub mniej zaawansowanych rozkładach. Zyski są szczególnie widoczne w równoważeniu poprawnego wykrywania eksonów z unikaniem fałszywych alarmów.
Co to oznacza dla analiz genomowych
Dla czytelników główny wniosek jest taki, że autorzy zbudowali bardziej precyzyjne „urządzenie nasłuchujące” genomu. Poprzez staranne mapowanie DNA na liczby, śledzenie jego rytmów w krótkich oknach, rozdzielanie sygnału na czyste składniki i celowe usuwanie szumu, uzyskują znacznie ostrzejszy obraz położenia instrukcji kodujących białka. Chociaż obecna implementacja może być obliczeniowo wymagająca i wciąż wymaga dostrajania niektórych ustawień, ramy te pokazują, że narzędzia nowoczesnego przetwarzania sygnałów mogą znacząco poprawić odczyt genomu. W dłuższej perspektywie takie metody mogą pomóc naukowcom szybciej adnotować nowe genomy i wspierać dalsze badania funkcji genów, mechanizmów chorób oraz medycyny spersonalizowanej.
Cytowanie: Benarjee, S., Vaegae, N.K. A STDFT-CEEMD approach with wavelet packet thresholding for exon prediction in eukaryotic cells. Sci Rep 16, 15948 (2026). https://doi.org/10.1038/s41598-026-43722-2
Słowa kluczowe: predykcja eksonów, genomiczne przetwarzanie sygnałów, analiza DNA, obszary kodujące białka, redukcja szumów