Clear Sky Science · fr
Une approche STDFT-CEEMD avec seuillage par paquets d’ondelettes pour la prédiction des exons dans les cellules eucaryotes
Identifier les parties utiles de notre code génétique
À l’intérieur de chaque cellule, de longues chaînes d’ADN portent des instructions pour fabriquer les protéines qui nous maintiennent en vie. Mais seules certaines portions de cet ADN codent effectivement pour des protéines, tandis que de larges segments jouent plutôt un rôle de ponctuation ou de toile de fond. Cet article s’attaque à un défi central de la génétique moderne : comment repérer de manière fiable les fragments codant pour des protéines, appelés exons, au sein d’un énorme volume de données brutes d’ADN en utilisant des outils de traitement du signal empruntés à l’ingénierie.
Pourquoi séparer le signal du bruit est important
Les gènes chez l’humain et d’autres organismes complexes sont morcelés en exons, qui portent les instructions utiles, et en introns, qui n’en portent pas. Lors de la production des protéines, les cellules copient l’ADN en ARN puis éliminent les introns, reliant les exons pour former un message final qui détermine la composition de la protéine. Identifier où commencent et se terminent les exons est crucial pour comprendre le fonctionnement des gènes, l’apparition des maladies et l’ajustement des traitements. Les méthodes informatiques traditionnelles reposent souvent sur de larges jeux de données étiquetées ou sur des modèles biologiques détaillés, qui ne sont pas toujours disponibles ou peuvent échouer pour des espèces peu étudiées. C’est pourquoi les méthodes pouvant travailler directement sur l’ADN brut, en le traitant comme un signal à analyser, sont de plus en plus attrayantes.
Transformer l’ADN en signal
Dans cette étude, les auteurs traitent l’ADN comme une forme d’onde, à l’instar d’une piste audio, puis appliquent une série d’étapes de traitement. D’abord, chacune des quatre lettres de l’ADN est convertie en nombres selon un schéma basé sur des matrices d’Hadamard, des motifs choisis de plus et moins un. Cette étape crée quatre pistes numériques distinctes qui conservent toute l’information de la séquence originale mais sont mieux adaptées à l’analyse. Ensuite, la méthode parcourt la séquence avec une fenêtre glissante et utilise un outil temps–fréquence appelé transformée de Fourier discrète à court terme (Short-Time Discrete Fourier Transform) pour rechercher un motif répétitif apparaissant toutes les trois bases. Ce rythme « périodicité-3 » est une caractéristique connue des régions codantes, car les protéines sont construites à partir de mots de trois lettres, ou codons, dans le code génétique.
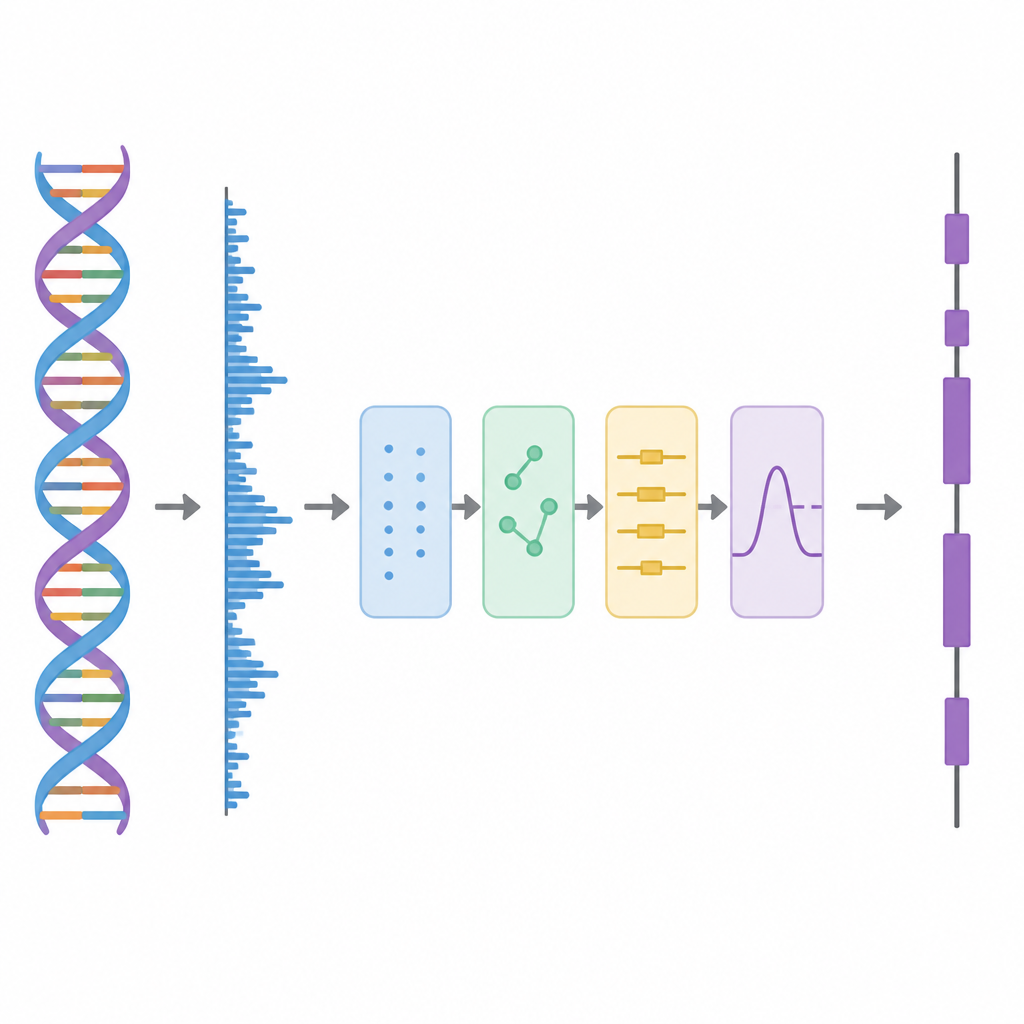
Décomposer les couches du signal
Les données génomiques réelles sont désordonnées. Les tendances de fond à longue portée et les fluctuations aléatoires peuvent estomper le motif périodicité-3, en particulier pour les exons courts. Pour y remédier, les auteurs empruntent une idée de la décomposition avancée des signaux, où une forme d’onde complexe est scindée en composantes plus simples. Ils utilisent une technique appelée Complete Ensemble Empirical Mode Decomposition (CEEMD), qui ajoute à plusieurs reprises un bruit soigneusement calibré puis moyenne les résultats pour produire un ensemble de composantes plus propres. Une mesure d’autocorrélation est ensuite utilisée pour décider lesquelles de ces composantes contiennent une structure significative et lesquelles sont dominées par le bruit. Les composantes bruitées sont ensuite nettoyées à l’aide d’un seuillage par paquets d’ondelettes, méthode qui supprime les petites variations saccadées tout en préservant la forme principale du signal.
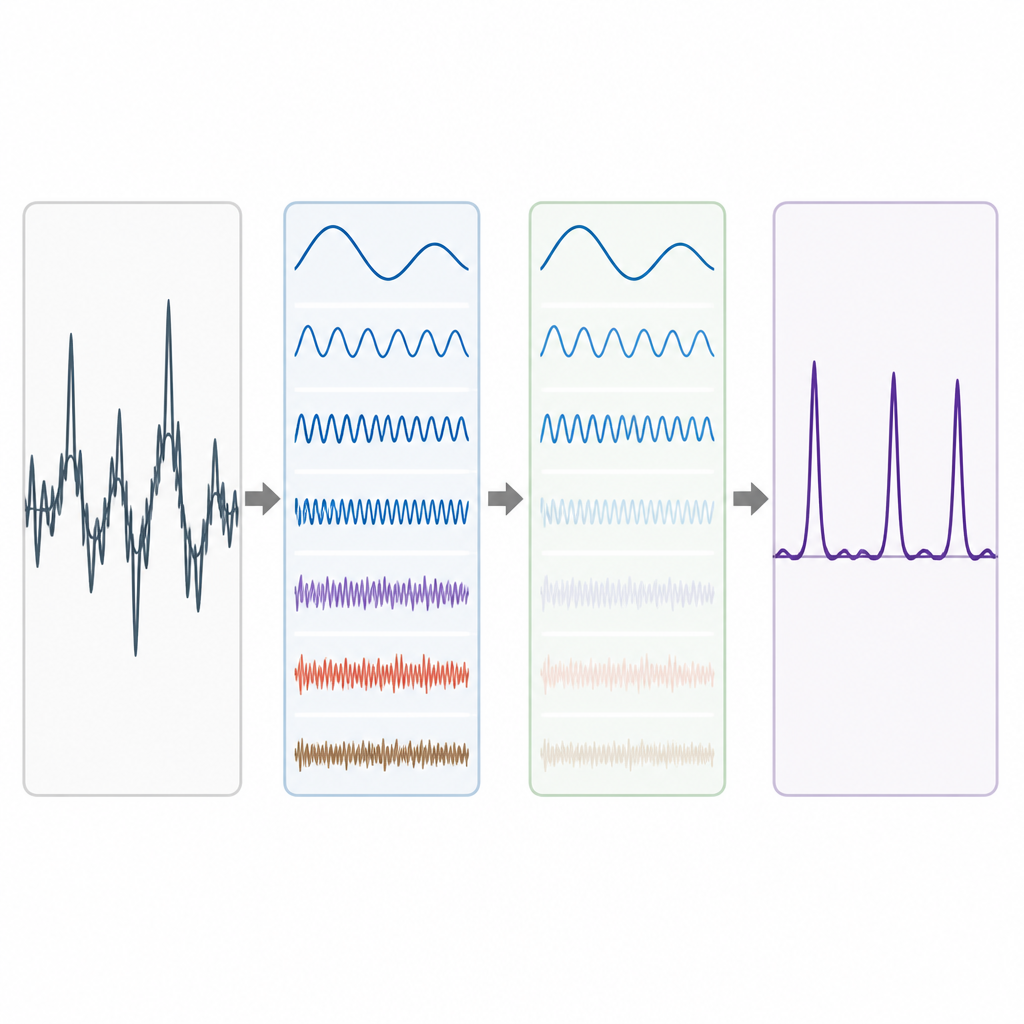
Tester la méthode sur des gènes réels
Pour évaluer l’efficacité de leur chaîne de traitement, les auteurs l’appliquent à des gènes bien étudiés du nématode Caenorhabditis elegans et de la souris, ainsi qu’à une collection de référence de 195 segments de gènes humains, murins et rat. Dans chaque cas, ils comparent leurs prédictions d’exons avec des annotations d’experts. Leur approche produit des pics plus nets aux emplacements des vrais exons et un niveau de fond plus bas dans les régions non codantes. Lorsqu’ils synthétisent les performances à l’aide de mesures courantes telles que la sensibilité, la spécificité, la précision et l’aire sous la courbe ROC, leur méthode surpasse de manière constante plusieurs approches antérieures de traitement du signal reposant sur des filtres plus simples ou des décompositions moins raffinées. Les gains sont particulièrement notables pour l’équilibre entre la détection correcte des exons et la limitation des fausses alertes.
Ce que cela change pour l’analyse génomique
Pour le lecteur, l’idée principale est que les auteurs ont construit un « dispositif d’écoute » plus précis pour le génome. En cartographiant soigneusement l’ADN en nombres, en suivant ses rythmes sur de courtes fenêtres, en décomposant le signal en composantes propres et en éliminant le bruit de façon ciblée, ils obtiennent une image beaucoup plus nette des endroits où se trouvent les instructions codantes pour les protéines. Bien que l’implémentation actuelle puisse être gourmande en calcul et nécessite encore le réglage de certains paramètres, le cadre montre que les outils du traitement moderne du signal peuvent améliorer significativement notre façon de lire le génome. À long terme, de telles méthodes pourraient aider les scientifiques à annoter de nouveaux génomes plus rapidement et soutenir des études ultérieures sur la fonction des gènes, les mécanismes des maladies et la médecine personnalisée.
Citation: Benarjee, S., Vaegae, N.K. A STDFT-CEEMD approach with wavelet packet thresholding for exon prediction in eukaryotic cells. Sci Rep 16, 15948 (2026). https://doi.org/10.1038/s41598-026-43722-2
Mots-clés: prédiction d’exons, traitement du signal génomique, analyse de l’ADN, régions codantes en protéines, réduction du bruit