Clear Sky Science · pt
Uma abordagem STDFT-CEEMD com limiarização por pacotes wavelet para predição de éxons em células eucarióticas
Encontrando as Partes Úteis do Nosso Código Genético
Dentro de cada célula, longas cadeias de DNA carregam instruções para construir as proteínas que nos mantêm vivos. Mas apenas certos trechos desse DNA realmente codificam proteínas, enquanto grandes seções funcionam mais como pontuação ou plano de fundo. Este artigo aborda um desafio central da genética moderna: como detectar de maneira confiável trechos codificadores de proteínas, chamados éxons, em grandes volumes de dados brutos de DNA usando ferramentas inteligentes de processamento de sinais emprestadas da engenharia.
Por que Separar Sinal de Ruído Importa
Genes em humanos e outros organismos complexos são fragmentados em éxons, que carregam instruções úteis, e íntrons, que não codificam. Durante a produção de proteínas, as células copiam o DNA para RNA e então removem os íntrons, costurando os éxons em uma mensagem final que determina a composição da proteína. Identificar onde os éxons começam e terminam é crucial para entender como os genes funcionam, como surgem doenças e como tratamentos podem ser personalizados. Métodos tradicionais dependem fortemente de grandes conjuntos de dados rotulados ou de modelos biológicos detalhados, que nem sempre estão disponíveis ou podem falhar em espécies pouco estudadas. Por isso, métodos que operam diretamente sobre o DNA bruto, tratando-o como um sinal a ser analisado, são cada vez mais atraentes.
Transformando DNA em um Sinal
Neste estudo, os autores tratam o DNA como se fosse uma forma de onda, similar a uma faixa de áudio, e aplicam então uma sequência de etapas de processamento. Primeiro, cada uma das quatro letras do DNA é convertida em números usando um esquema especial baseado em matrizes de Hadamard, que são padrões cuidadosamente escolhidos de mais e menos uns. Essa etapa cria quatro trilhas numéricas limpas que preservam todas as informações da sequência original, mas ficam mais adequadas para análise. Em seguida, o método varre a sequência com uma janela deslizante e usa uma ferramenta tempo–frequência chamada Transformada Discreta de Fourier de Curto Tempo para buscar um padrão repetitivo que aparece a cada três bases. Esse ritmo de “período 3” é uma característica bem conhecida das regiões codificadoras porque as proteínas são construídas a partir de palavras de três letras, ou códons, no código genético.
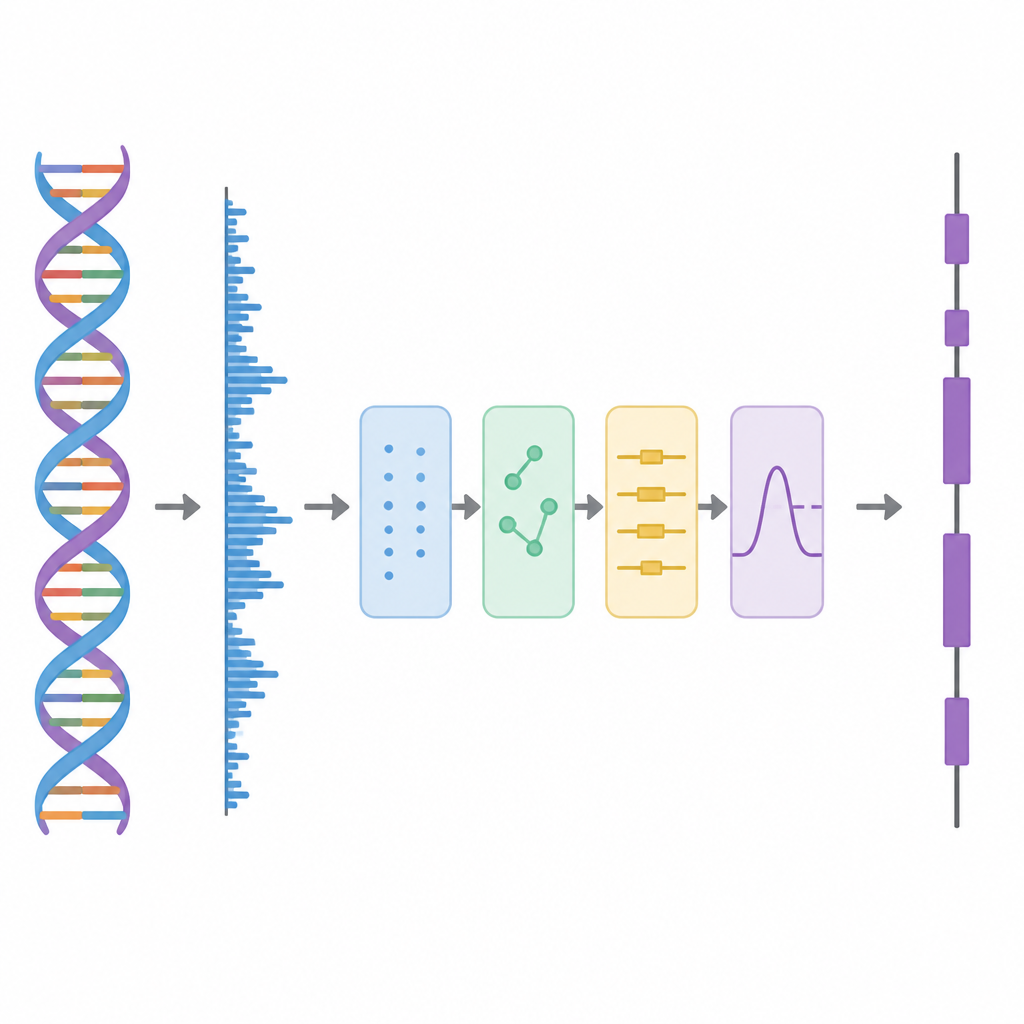
Descascar as Camadas do Sinal
Dados genômicos reais são bagunçados. Tendências de fundo de longo alcance e flutuações aleatórias podem borrar o padrão de período 3, especialmente em éxons curtos. Para enfrentar isso, os autores adotam uma ideia da decomposição avançada de sinais, na qual uma forma de onda complicada é dividida em blocos construtivos mais simples. Eles usam uma técnica chamada Decomposição Empírica de Modo com Conjunto Completo (CEEMD), que adiciona repetidamente ruído balanceado e então faz a média dos resultados para produzir um conjunto de componentes mais limpos. Uma medida de autocorrelação é então usada para decidir quais desses componentes carregam estrutura significativa e quais são dominados pelo ruído. As partes ruidosas são então limpas adicionalmente usando limiarização por pacotes wavelet, um método que elimina pequenas variações tremidas enquanto preserva a forma principal do sinal.
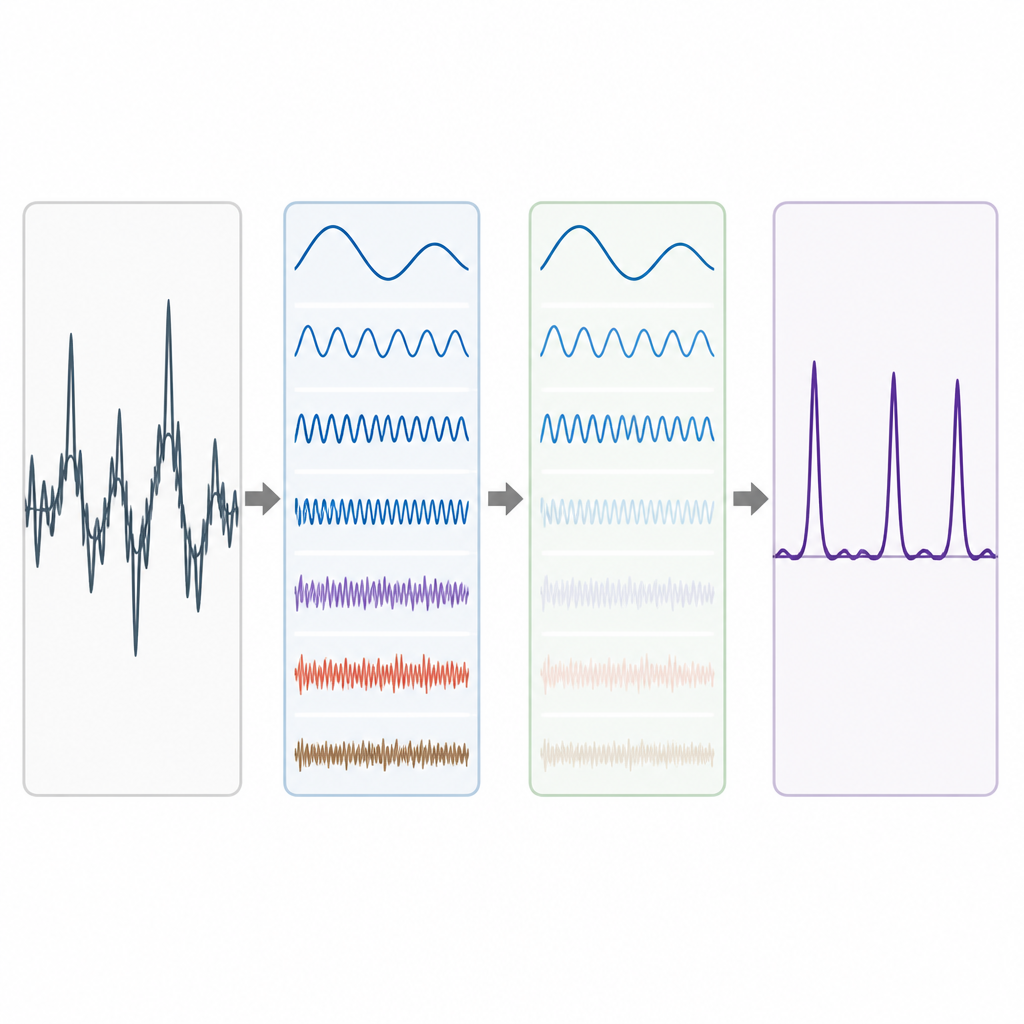
Testando o Método em Genes Reais
Para avaliar o desempenho do pipeline, os autores o aplicam a genes bem estudados do nematoide Caenorhabditis elegans e do camundongo doméstico, assim como a uma coleção de referência de 195 segmentos gênicos de humano, camundongo e rato. Em cada caso, eles comparam suas predições de éxons com anotações de especialistas. A abordagem produz picos mais claros onde éxons verdadeiros ocorrem e menor nível de fundo em regiões que não codificam proteínas. Ao resumir o desempenho com medidas comuns como sensibilidade, especificidade, acurácia e área sob a curva ROC, o método supera consistentemente várias abordagens anteriores de processamento de sinais que dependem de filtros mais simples ou decomposições menos refinadas. Os ganhos são especialmente notáveis no equilíbrio entre a detecção correta de éxons e a redução de falsos positivos.
O Que Isso Significa para a Análise Genômica
Para o leitor, a principal conclusão é que os autores construíram um “aparelho de escuta” mais preciso para o genoma. Ao mapear cuidadosamente o DNA em números, acompanhar seus ritmos em janelas curtas, separar o sinal em componentes limpos e remover o ruído de forma direcionada, eles obtêm uma visão muito mais nítida de onde residem as instruções codificadoras de proteínas. Embora a implementação atual possa ser computacionalmente pesada e ainda exija ajuste de certos parâmetros, o arcabouço mostra que ferramentas do processamento moderno de sinais podem melhorar de modo significativo como lemos o genoma. A longo prazo, esses métodos podem ajudar cientistas a anotar novos genomas mais rapidamente e apoiar estudos posteriores sobre função gênica, mecanismos de doença e medicina personalizada.
Citação: Benarjee, S., Vaegae, N.K. A STDFT-CEEMD approach with wavelet packet thresholding for exon prediction in eukaryotic cells. Sci Rep 16, 15948 (2026). https://doi.org/10.1038/s41598-026-43722-2
Palavras-chave: predição de éxons, processamento de sinais genômicos, análise de DNA, regiões codificadoras de proteína, redução de ruído