Clear Sky Science · de
Ein STDFT-CEEMD-Ansatz mit Wavelet-Paket-Schwellenwertbildung zur Exon-Vorhersage in eukaryotischen Zellen
Die nützlichen Teile unseres genetischen Codes finden
In jeder Zelle tragen lange DNA-Stränge die Anweisungen zum Aufbau der Proteine, die unser Leben ermöglichen. Nur bestimmte Abschnitte dieser DNA kodieren tatsächlich Proteine, während große Teile eher wie Interpunktion oder Hintergrund wirken. Diese Arbeit adressiert eine zentrale Herausforderung der modernen Genetik: Wie erkennt man zuverlässig die protein-kodierenden Abschnitte, sogenannte Exons, in riesigen Mengen roher DNA-Daten mithilfe intelligenter, aus der Ingenieurwissenschaft entliehener Signalverarbeitungswerkzeuge?
Warum die Trennung von Signal und Rauschen wichtig ist
Gene von Menschen und anderen komplexen Organismen sind in Exons, die nützliche Informationen tragen, und Introns, die dies nicht tun, gegliedert. Während der Proteinsynthese kopieren Zellen DNA in RNA und schneiden dann die Introns heraus, wobei die Exons zu einer finalen Botschaft zusammengesetzt werden, die den Aufbau eines Proteins bestimmt. Die Identifikation der Exon-Grenzen ist entscheidend, um zu verstehen, wie Gene funktionieren, wie Krankheiten entstehen und wie Therapien angepasst werden können. Klassische Rechenverfahren sind stark auf umfangreiche, sorgfältig beschriftete Trainingsdaten oder detaillierte biologische Modelle angewiesen, die nicht immer verfügbar sind oder bei wenig untersuchten Arten versagen können. Daher sind Methoden, die direkt auf roher DNA arbeiten und diese wie ein zu analysierendes Signal behandeln, zunehmend attraktiv.
DNA in ein Signal verwandeln
In dieser Studie behandeln die Autoren DNA wie eine Wellenform, ähnlich einer Audiospur, und wenden dann eine Folge von Verarbeitungsschritten an. Zunächst wird jeder der vier DNA-Buchstaben mithilfe eines speziellen Schemas auf Hadamard-Matrizenbasis in Zahlen überführt, das aus sorgfältig gewählten Mustern von Plus- und Minus-Einsen besteht. Dieser Schritt erzeugt vier saubere numerische Spuren, die alle Informationen der ursprünglichen Sequenz bewahren, aber besser für die Analyse geeignet sind. Anschließend durchsucht die Methode die Sequenz mit einem gleitenden Fenster und nutzt ein Zeit-Frequenz-Werkzeug, die Short-Time Discrete Fourier Transform (STDFT), um nach einem sich alle drei Basen wiederholenden Muster zu suchen. Dieser „Period-3“-Rhythmus ist ein bekanntes Merkmal protein-kodierender Regionen, weil Proteine aus drei-Basen-Wörtern, den Codons, aufgebaut werden.
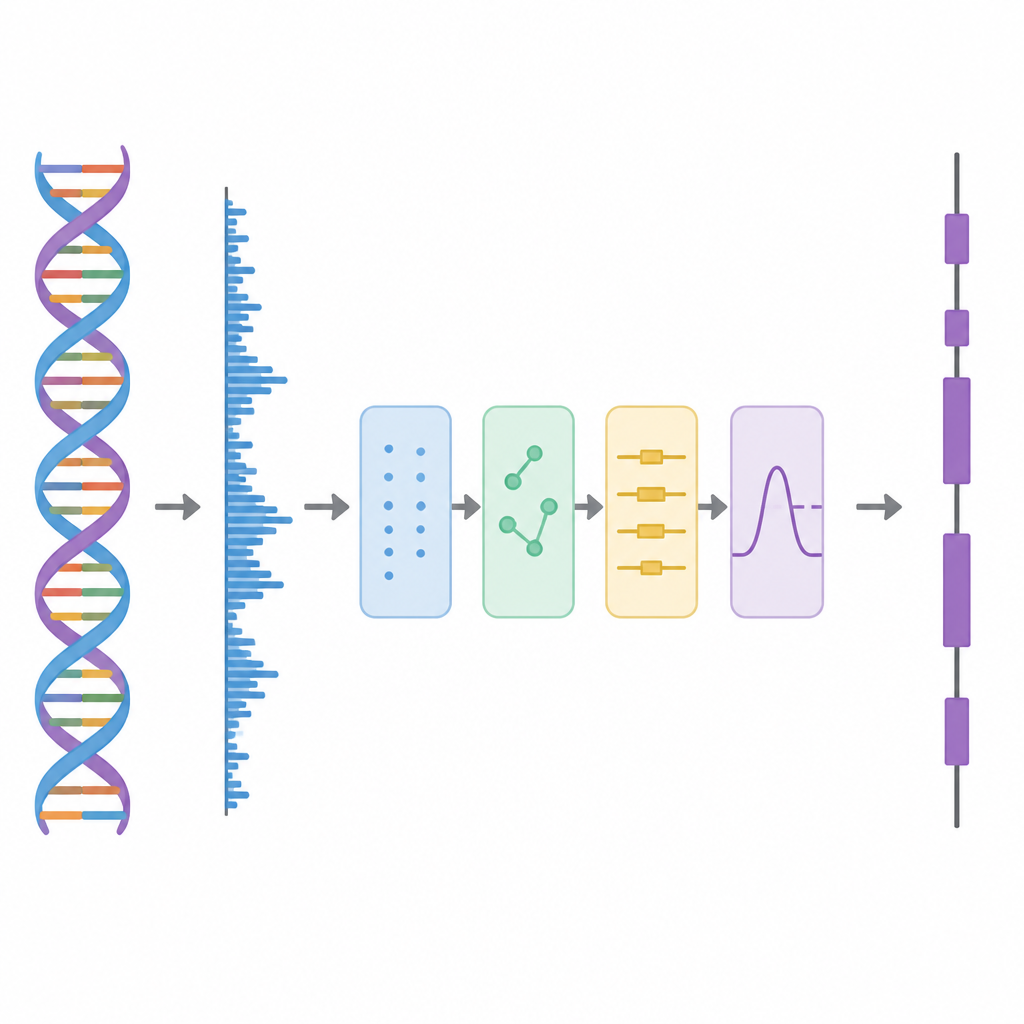
Die Schichten des Signals auseinanderziehen
Echte genomische Daten sind unordentlich. Langfristige Hintergrundtrends und zufällige Schwankungen können das Period-3-Muster verwischen, besonders bei kurzen Exons. Um dem zu begegnen, übernehmen die Autoren eine Idee aus der fortgeschrittenen Signalzerlegung, bei der eine komplexe Wellenform in einfachere Bausteine zerlegt wird. Sie verwenden eine Technik namens Complete Ensemble Empirical Mode Decomposition (CEEMD), die wiederholt sorgfältig ausbalanciertes Rauschen hinzufügt und die Ergebnisse mittelt, um eine Menge sauberer Komponenten zu erzeugen. Eine Selbstkorrelationsmaßzahl wird dann verwendet, um zu entscheiden, welche dieser Komponenten sinnvolle Struktur tragen und welche vom Rauschen dominiert sind. Die verrauschten Teile werden anschließend mit Wavelet-Paket-Schwellenwertbildung gereinigt, einer Methode, die kleine, zittrige Variationen entfernt und dabei die Hauptform des Signals bewahrt.
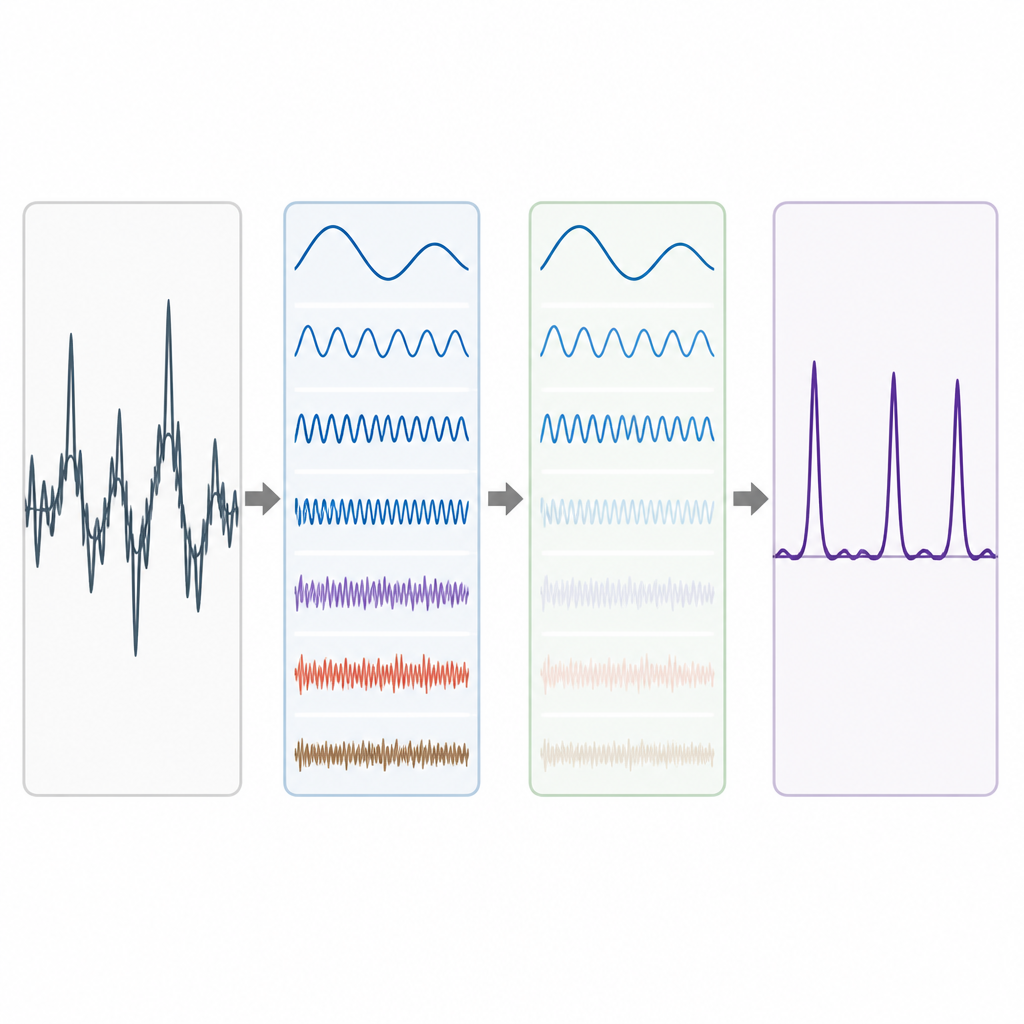
Die Methode an echten Genen testen
Um zu prüfen, wie gut ihre Pipeline funktioniert, wenden die Autoren sie auf gut untersuchte Gene des Fadenwurms Caenorhabditis elegans und der Hausmaus sowie auf eine Benchmark-Sammlung von 195 Genabschnitten aus Mensch, Maus und Ratte an. In jedem Fall vergleichen sie ihre Exon-Vorhersagen mit Expertenannotationen. Ihr Ansatz erzeugt klarere Spitzen an Stellen, an denen tatsächlich Exons vorkommen, und einen geringeren Hintergrund in Regionen, die keine Proteine kodieren. Fasst man die Leistung mit gängigen Maßen wie Sensitivität, Spezifität, Genauigkeit und Fläche unter der ROC-Kurve zusammen, übertrifft ihre Methode wiederholt mehrere frühere signalverarbeitungstechnische Ansätze, die auf einfacheren Filtern oder weniger ausgefeilten Zerlegungen beruhen. Die Verbesserungen sind besonders bemerkbar beim Ausgleich zwischen korrekter Exon-Erkennung und der Vermeidung von Fehlalarmen.
Was das für die Genomanalyse bedeutet
Für die Leserschaft ist die wichtigste Erkenntnis, dass die Autoren ein präziseres „Abhörgerät“ für das Genom entwickelt haben. Durch das sorgfältige Abbilden von DNA in Zahlen, das Nachverfolgen ihrer Rhythmen über kurze Fenster, das Auseinanderziehen des Signals in saubere Komponenten und das gezielte Entfernen von Rauschen erhalten sie einen deutlich schärferen Blick darauf, wo protein-kodierende Anweisungen liegen. Obwohl die aktuelle Implementierung rechenintensiv sein kann und noch das Abstimmen bestimmter Einstellungen erfordert, zeigt das Framework, dass Werkzeuge der modernen Signalverarbeitung die Art und Weise, wie wir das Genom lesen, erheblich verbessern können. Langfristig könnten solche Methoden Wissenschaftlern helfen, neue Genome schneller zu annotieren und nachgelagerte Studien zu Genfunktion, Krankheitsmechanismen und personalisierter Medizin zu unterstützen.
Zitation: Benarjee, S., Vaegae, N.K. A STDFT-CEEMD approach with wavelet packet thresholding for exon prediction in eukaryotic cells. Sci Rep 16, 15948 (2026). https://doi.org/10.1038/s41598-026-43722-2
Schlüsselwörter: Exon-Vorhersage, genomische Signalverarbeitung, DNA-Analyse, protein-kodierende Regionen, Rauschunterdrückung