Clear Sky Science · es
Un enfoque STDFT-CEEMD con umbralización por paquetes de wavelets para la predicción de exones en células eucariotas
Encontrar las partes útiles de nuestro código genético
Dentro de cada célula, largas cadenas de ADN contienen instrucciones para construir las proteínas que nos mantienen con vida. Pero solo ciertos tramos de este ADN codifican realmente proteínas, mientras que secciones extensas actúan más como puntuación o fondo. Este artículo aborda un reto clave de la genética moderna: cómo identificar con fiabilidad las piezas codificantes de proteínas, llamadas exones, dentro de ingentes cantidades de datos brutos de ADN usando herramientas inteligentes de procesamiento de señales tomadas de la ingeniería.
Por qué importa separar señal de ruido
Los genes en humanos y otros organismos complejos están fragmentados en exones, que portan instrucciones útiles, e intrones, que no. Durante la producción de proteínas, las células copian el ADN en ARNm y luego recortan los intrones, cosiendo los exones entre sí para formar el mensaje final que determina la composición de una proteína. Identificar dónde comienzan y terminan los exones es crucial para entender cómo funcionan los genes, cómo surgen las enfermedades y cómo se podrían adaptar los tratamientos. Los métodos informáticos tradicionales dependen en gran medida de grandes conjuntos de datos anotados o de modelos biológicos detallados, que no siempre están disponibles o pueden fallar en especies poco estudiadas. Por eso son cada vez más atractivos los métodos que pueden trabajar directamente sobre el ADN bruto, tratándolo como una señal a analizar.
Convertir el ADN en una señal
En este estudio, los autores tratan el ADN como si fuese una forma de onda, similar a una pista de audio, y aplican a continuación una serie de pasos de procesamiento. Primero, cada una de las cuatro letras del ADN se convierte en números mediante un esquema especial basado en matrices de Hadamard, que son patrones cuidadosamente escogidos de unos y menos unos. Este paso crea cuatro pistas numéricas limpias que conservan toda la información de la secuencia original pero son más adecuadas para el análisis. A continuación, el método recorre la secuencia con una ventana deslizante y utiliza una herramienta tiempo‑frecuencia llamada Transformada Discreta de Fourier de Tiempo Corto para buscar un patrón repetitivo que aparece cada tres bases. Este ritmo de “periodicidad 3” es una característica bien conocida de las regiones codificantes porque las proteínas se construyen a partir de palabras de tres letras, o codones, en el código genético.
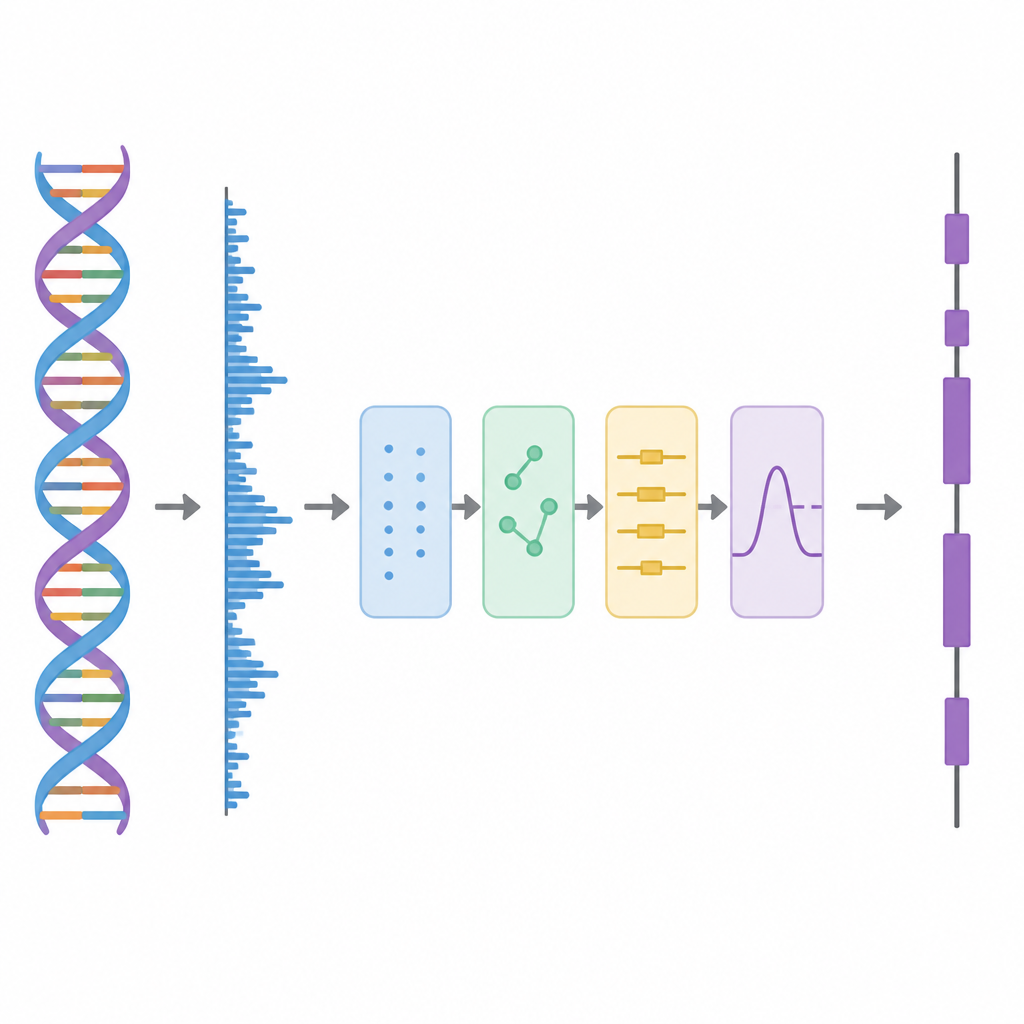
Despejar las capas de la señal
Los datos genómicos reales son desordenados. Las tendencias de fondo a largo plazo y las fluctuaciones aleatorias pueden difuminar el patrón de periodicidad 3, especialmente en exones cortos. Para abordar esto, los autores recurren a una idea de la descomposición avanzada de señales, en la que una forma de onda complicada se divide en bloques constructivos más simples. Usan una técnica llamada Descomposición Empírica en Conjunto Completo (Complete Ensemble Empirical Mode Decomposition), que añade repetidamente ruido cuidadosamente balanceado y luego promedia los resultados para producir un conjunto de componentes más limpias. A continuación, se utiliza una medida de autocorrelación para decidir cuáles de estos componentes contienen estructura significativa y cuáles están dominados por ruido. Las piezas ruidosas se limpian más mediante umbralización con paquetes de wavelets, un método que recorta variaciones pequeñas y temblorosas mientras preserva la forma principal de la señal.
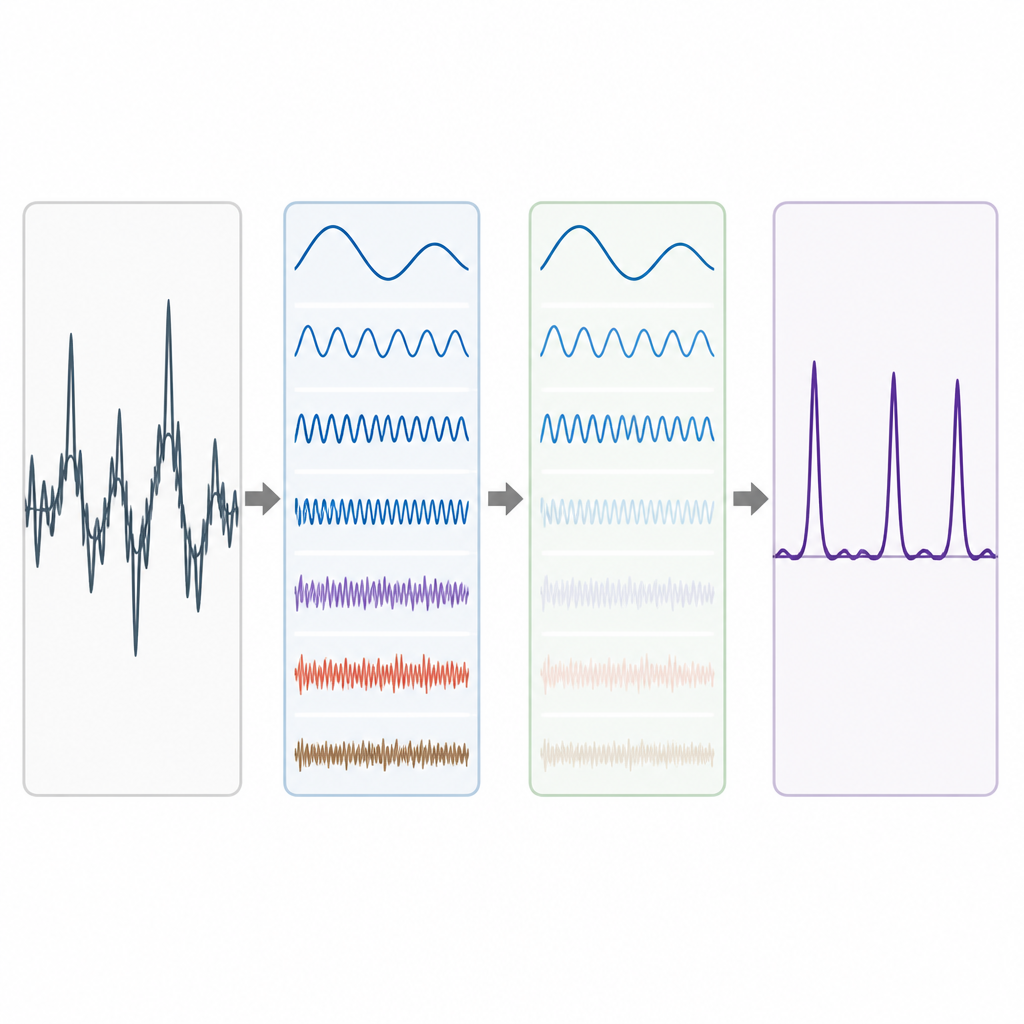
Probar el método en genes reales
Para evaluar qué tan bien funciona su flujo de trabajo, los autores lo aplican a genes bien estudiados del nematodo Caenorhabditis elegans y del ratón doméstico, así como a una colección de referencia de 195 segmentos génicos humanos, de ratón y de rata. En cada caso comparan sus predicciones de exones con anotaciones de expertos. Su enfoque produce picos más nítidos donde se encuentran los exones verdaderos y menor nivel de fondo en regiones que no codifican proteínas. Al resumir el rendimiento con medidas comunes como sensibilidad, especificidad, precisión y área bajo la curva ROC, su método supera de forma constante a varios enfoques previos de procesamiento de señales que dependen de filtros más simples o descomposiciones menos refinadas. Las mejoras son especialmente notables al equilibrar la detección correcta de exones con la evitación de falsas alarmas.
Qué significa esto para el análisis genómico
Para el lector, la conclusión principal es que los autores han construido un “dispositivo de escucha” más preciso para el genoma. Al mapear cuidadosamente el ADN a números, rastrear sus ritmos en ventanas cortas, descomponer la señal en componentes limpias y eliminar el ruido de forma dirigida, obtienen una visión mucho más nítida de dónde residen las instrucciones codificantes de proteínas. Aunque la implementación actual puede ser costosa en cómputo y aún requiere ajustar ciertos parámetros, el marco muestra que las herramientas del procesamiento moderno de señales pueden mejorar significativamente la lectura del genoma. A largo plazo, tales métodos podrían ayudar a los científicos a anotar nuevos genomas más rápido y apoyar estudios posteriores sobre función génica, mecanismos de enfermedad y medicina personalizada.
Cita: Benarjee, S., Vaegae, N.K. A STDFT-CEEMD approach with wavelet packet thresholding for exon prediction in eukaryotic cells. Sci Rep 16, 15948 (2026). https://doi.org/10.1038/s41598-026-43722-2
Palabras clave: predicción de exones, procesamiento de señales genómicas, análisis de ADN, regiones codificantes de proteínas, reducción de ruido