Clear Sky Science · sv
Ny universell domäncentrerad metod för proteinklassificering
Varför det är viktigt att sortera proteiner för hälsan
Inuti varje cell håller tusentals små proteinmaskiner livet igång. Bland de viktigaste finns proteinkinaser, enzymer som slår på eller av andra proteiner och som är centrala mål för många moderna läkemedel, särskilt cancerterapier. Men forskare har fortfarande svårt att prydligt dela in alla kinaser i familjer som speglar hur de fungerar. Denna artikel presenterar ett nytt sätt att klassificera kinaser—och i princip många andra proteiner—genom att fokusera på den gemensamma kärnregionen som utför arbetet och på de grundläggande fysikalisk-kemiska egenskaperna hos dess byggstenar. Det lovar skarpare kartor över proteinfamiljer och i slutändan bättre ledtrådar för läkemedelsdesign.
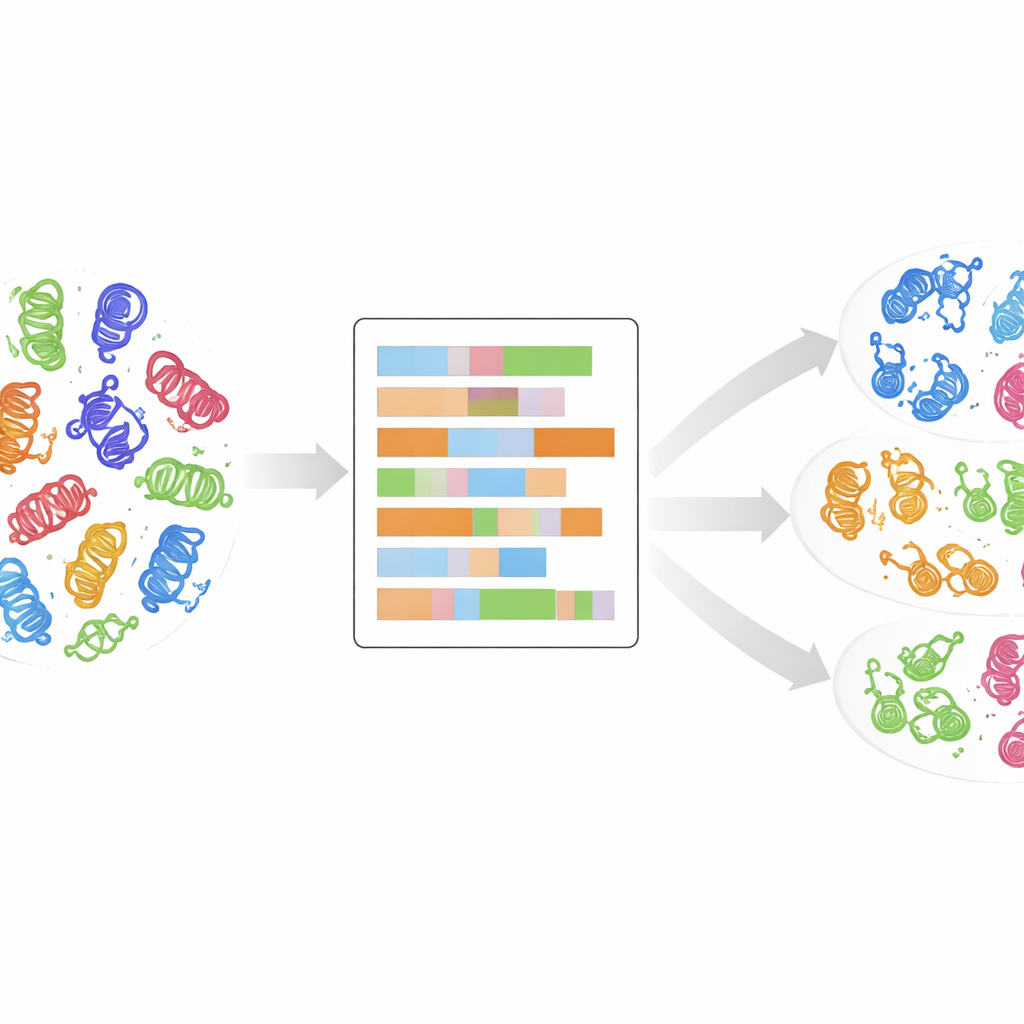
Från familjeträd till ett mer detaljerat fingeravtryck
Traditionellt har kinaser grupperats i klasser genom att jämföra deras gen- eller proteinsekvenser och bygga evolutionära ”familjeträd”. Denna metod har varit mycket framgångsrik och avslöjat cirka 500 mänskliga kinaser som delas in i flera huvudgrupper såsom AGC, CAMK, CMGC, STE, TK, TKL med flera. Ändå passar många nyupptäckta kinaser inte rent in i dessa grupper: deras sekvenser ser annorlunda ut, även om de kan bete sig liknande i celler. Standardjämförelser tar också lite hänsyn till de underliggande egenskaperna hos varje aminosyra—storlek, laddning eller vattenälskande karaktär som bestämmer hur ett protein veckas och fungerar. Författarna argumenterar för att för att verkligen förstå kinasfamiljer måste vi titta bortom alfabetliknande sekvensmatchning och undersöka dessa fysikalisk-kemiska fingeravtryck.
Zooma in på kinasernas aktiva kärna
För att göra detta använde forskarna en högkvalitativ inriktning av 497 mänskliga kinasdomäner, de kompakta kärnorna som faktiskt utför den kemiska reaktionen att tillsätta fosfatgrupper. För varje position i denna delade domänkarta ersatte de aminosyra-bokstaven med upp till 30 numeriska beskrivare som fångar egenskaper såsom laddning, hydrofobicitet, polaritet och storlek, plus en extra markör för gap. Resultatet är ett detaljerat numeriskt porträtt av varje kinasdomän, där liknande beteende i tredimensionellt utrymme bör översättas till liknande mönster i dessa tal. De reducerade sedan komplexiteten i dessa porträtt med hjälp av principal component analysis, en vedertagen teknik som kondenserar många mätvärden till några huvudriktningar som fångar de största skillnaderna.
Låta data bilda kluster själva
Utan att tala om för datorn vilken kinas som tillhörde vilken känd klass, använde teamet en oövervakad klustringsmetod kallad k-means på de reducerade numeriska data. De utforskade många möjliga antal kluster och använde statistiska mått för att identifiera de mest meningsfulla grupperingarna, och kombinerade sedan flera sådana lösningar till en slutlig uppsättning av 24 kluster, var och en med en konfidenspoäng som speglar hur stabilt det var över körningar. Anmärkningsvärt nog hamnade omkring 90 % av kinaserna i kluster som matchade deras ursprungliga klassetiketter, vilket tyder på att det fysikalisk-kemiska domänporträttet naturligt återhämtar—och ibland skärper—befintliga klassificeringar. Vissa kluster innehöll en blandning av en huvudklass och tidigare ”OTHER”-kinaser, vilket antyder att dessa avvikare faktiskt kan tillhöra en etablerad familj.
Upptäcka viktiga strukturella hetpunkter
Utöver gruppering avslöjar metoden vilka delar av kinasdomänen som faktiskt driver dessa skillnader. Genom att kombinera huvudkomponenterna med residuens egenskaper och sedan slumpa data i randomiseringstester, identifierade författarna specifika positioner vars egenskapsmönster starkt skiljer en klass från de andra. Ett utmärkande exempel är en plats i aktiveringsloopens region hos CMGC-kinaser som nästan alltid bär en positivt laddad rest, olik de flesta andra klasser. Strukturella modeller visar att i en representativ CMGC-kinas hjälper denna rest till att stabilisera närliggande fosforylerade platser som är avgörande för att slå på enzymet. Intressant nog delar en ”oklassificerad” kinas kallad CDC7 en liknande miljö vid denna plats, vilket stöder förutsägelsen att den beter sig som en CMGC-kinas även om dess evolutionära historia skiljer sig.
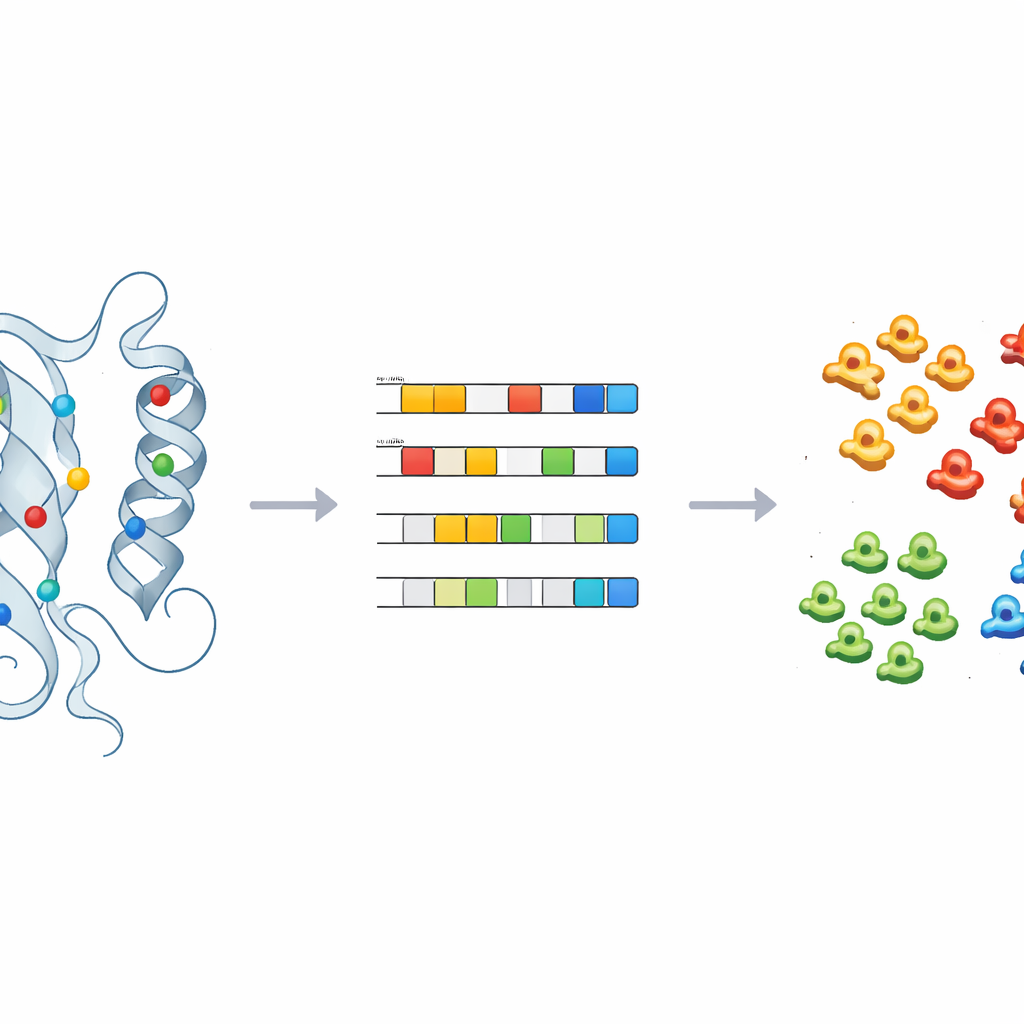
Lära maskiner att märka det okända
För att omsätta dessa insikter i praktiska förutsägelser tränade teamet övervakade maskininlärningsmodeller—inklusive logistisk regression, random forests och en probabilistisk klassificerare—på de fysikalisk-kemiska fingeravtrycken hos kinaser med kända etiketter. Efter noggrann justering och korsvalidering kunde dessa modeller med hög noggrannhet tilldela kinaser till huvudklasser med bara ett fåtal huvudkomponenter. Tillämpade på 66 kinaser som hade lämnats i catch-all-gruppen ”OTHER” tilldelade modellerna konsekvent flera, såsom CDC7 och medlemmar av ULK-familjen, till specifika kinas-klasser. Strukturella kontroller av dessa omklassificeringar, särskilt kring de nyckelpositioner som identifierats tidigare, stödde maskinernas förutsägelser och illustrerade hur metoden kan vägleda omklassificering och experimentell uppföljning.
Ett generellt recept för att kartlägga proteinfamiljer
I vardagliga termer visar detta arbete att proteiner kan sorteras inte bara genom att stava ut deras sekvenser, utan genom att destillera hur deras kärndelar beter sig fysiskt och kemiskt. För kinaser återfår denna domäncentrerade, egenskapsbaserade vy kända familjer, hjälper till att ommärka missanpassade exemplar och framhäver strukturella ”hetpunkter” som är viktiga för aktivitet och reglering. Eftersom receptet enbart bygger på en delad domäninriktning och generella aminosyra-beskrivare visar författarna också att det kan tillämpas på andra proteingrupper, såsom små GTPaser, och skulle kunna utsträckas till immunoglobuliner, G-proteinkopplade receptorer och vidare. När sådana kartor blir mer förfinade kan de styra designen av mer selektiva läkemedel, underlätta tolkningen av sjukdomsorsakande mutationer och erbjuda en klarare, funktionsfokuserad atlas över proteinuniversumet.
Citering: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Nyckelord: proteinkinaser, proteinklassificering, maskininlärning, proteindomäner, struktur-baserad biologi