Clear Sky Science · pl
Nowa uniwersalna metoda klasyfikacji białek skoncentrowana na domenach
Dlaczego porządkowanie białek ma znaczenie dla zdrowia
W każdej komórce tysiące drobnych maszyn białkowych utrzymują życie w działaniu. Do najważniejszych należą kinazy białkowe, enzymy włączające i wyłączające inne białka, będące głównymi celami wielu nowoczesnych leków, zwłaszcza w terapii nowotworów. Jednak naukowcy wciąż mają trudności z precyzyjnym przyporządkowaniem wszystkich kinaz do rodzin odzwierciedlających ich funkcję. Artykuł przedstawia nowy sposób klasyfikacji kinaz — i w zasadzie wielu innych białek — poprzez skoncentrowanie się na wspólnej, rdzeniowej domenie wykonującej pracę oraz na podstawowych właściwościach fizycznych i chemicznych jej elementów budulcowych. To obiecuje ostrzejsze mapy rodzin białkowych i — ostatecznie — lepsze wskazówki przy projektowaniu leków.
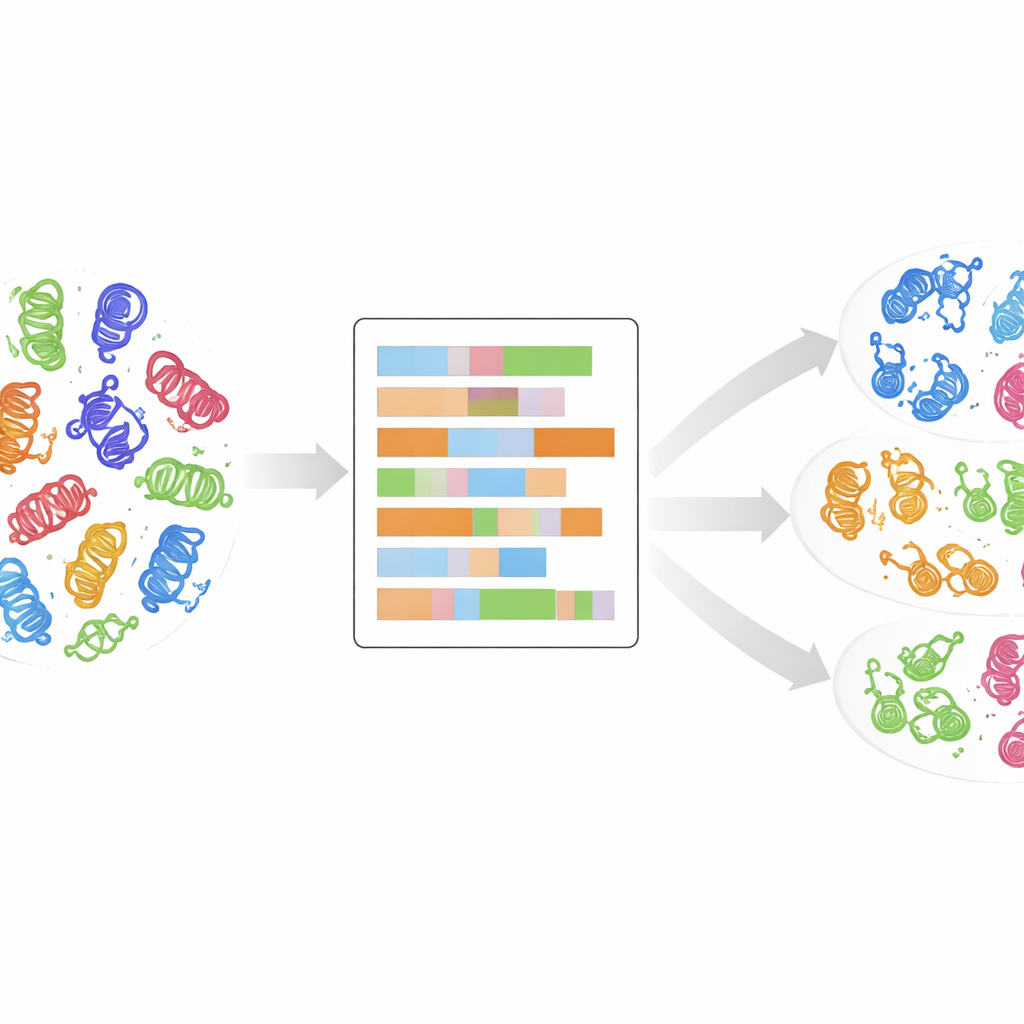
Od drzew rodowych do bardziej szczegółowego odcisku palca
Tradycyjnie kinazy grupowano według porównania ich sekwencji genów lub białek i budowania ewolucyjnych „drzew rodzinnych”. Podejście to odniosło duży sukces, ujawniając około 500 ludzkich kinaz, które dzielą się na kilka głównych grup, takich jak AGC, CAMK, CMGC, STE, TK, TKL i inne. Jednak wiele nowo odkrytych kinaz nie pasuje gładko do tych grup: ich sekwencje wyglądają inaczej, choć mogą zachowywać się podobnie w komórkach. Standardowe porównania zwracają też mało uwagi na podstawowe właściwości poszczególnych aminokwasów — rozmiar, ładunek czy hydrofilowość — które determinują składanie i funkcję białka. Autorzy argumentują, że aby naprawdę zrozumieć rodziny kinaz, trzeba wyjść poza dopasowywanie przypominające alfabet i przeanalizować te fizykochemiczne odciski palca.
Przybliżenie do aktywnego rdzenia kinaz
Aby to osiągnąć, badacze użyli wysokiej jakości wyrównania 497 ludzkich domen kinaz, kompaktowych rdzeni, które rzeczywiście przeprowadzają reakcję dodania grup fosforanowych. Dla każdej pozycji w tej wspólnej mapie domeny zastąpili literę aminokwasu aż do 30 numerycznymi deskryptorami opisującymi właściwości takie jak ładunek, hydrofobowość, polarność i rozmiar, oraz dodatkowym znacznikiem dla braków (gapów). Wynikiem jest szczegółowy numeryczny portret każdej domeny kinazy, w którym podobne zachowanie w trójwymiarowej przestrzeni powinno przekładać się na podobne wzorce tych liczb. Następnie zredukowali złożoność tych portretów za pomocą analizy głównych składowych (PCA), standardowej techniki kondensującej wiele pomiarów do kilku głównych kierunków wychwytujących największe różnice.
Pozwolenie danym na samogrupowanie
Bez informowania komputera, która kinaza należy do której znanej klasy, zespół zastosował nadzorowaną metodę grupowania zwaną k-means do zredukowanych danych numerycznych. Eksplorowali wiele możliwych liczebności klastrów i użyli statystycznych miar, by zidentyfikować najbardziej sensowne podziały, a następnie połączyli kilka takich rozwiązań w końcowy zbiór 24 klastrów, z których każdy otrzymał ocenę pewności odzwierciedlającą jego stabilność między uruchomieniami. Co ważne, około 90% kinaz trafiło do klastrów zgodnych z ich pierwotnymi etykietami klas, co sugeruje, że fizykochemiczny portret domeny naturalnie odtwarza — i czasem doprecyzowuje — istniejące klasyfikacje. Niektóre klastry zawierały mieszankę dominującej klasy i uprzednio oznaczonych jako „OTHER” kinaz, co sugeruje, że te odstające próbki mogą faktycznie należeć do ustalonej rodziny.
Odkrywanie kluczowych gorących miejsc strukturalnych
Ponad samym grupowaniem metoda ujawnia, które części domeny kinazy rzeczywiście napędzają te różnice. Łącząc główne składowe z właściwościami reszt i przeprowadzając testy losowania (randomizacji), autorzy wyodrębnili konkretne pozycje, których wzorce właściwości wyraźnie odróżniają jedną klasę od pozostałych. Przykładem jest miejsce w regionie pętli aktywacyjnej kinaz CMGC, które niemal zawsze zawiera dodatnio naładowaną resztę, w przeciwieństwie do większości innych klas. Modele strukturalne pokazują, że w reprezentatywnej kinazie CMGC ta reszta pomaga stabilizować pobliskie reszty fosforylowane, kluczowe dla aktywacji enzymu. Co ciekawe, kinaza nie sklasyfikowana wcześniej jako należąca do danej grupy — CDC7 — ma podobne środowisko w tym miejscu, co wspiera przewidywanie, że jej zachowanie zbliża się do kinaz CMGC, nawet jeśli jej historia ewolucyjna jest inna.
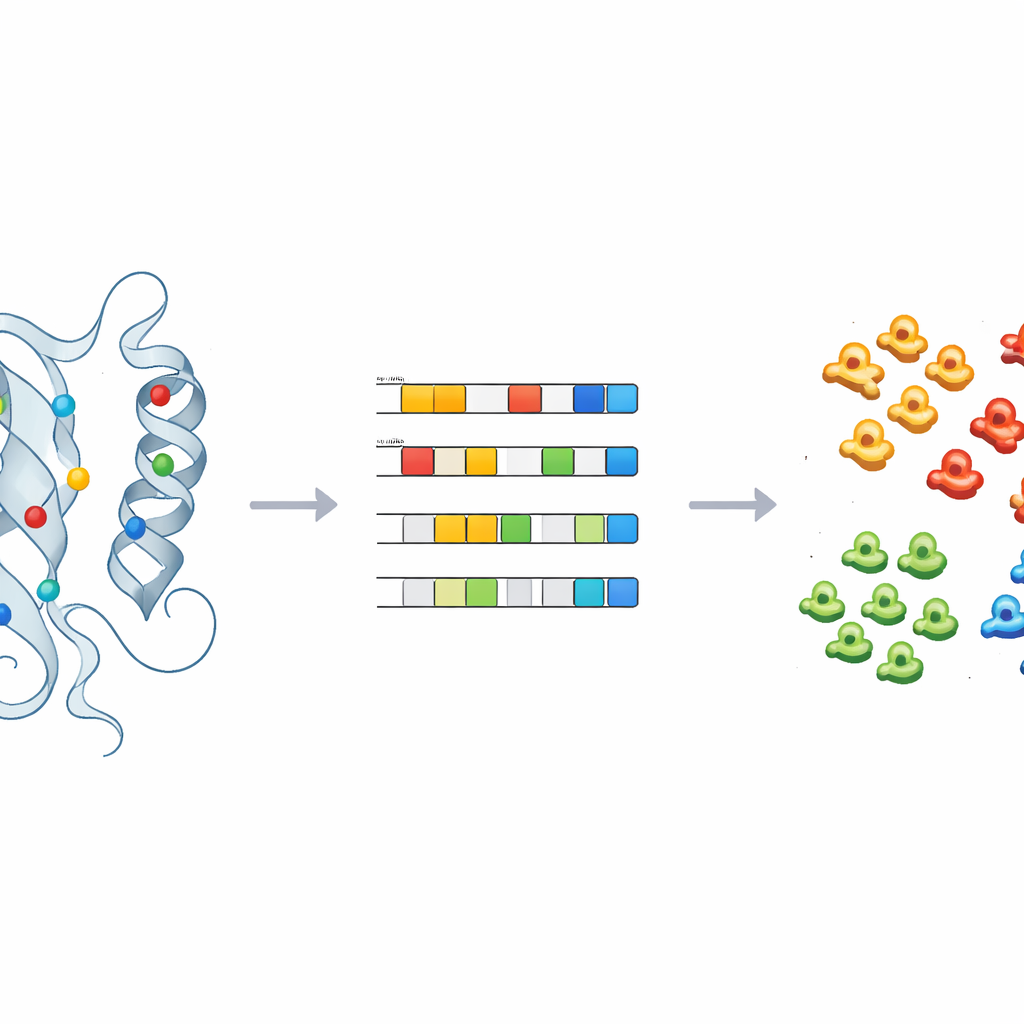
Nauczanie maszyn etykietowania nieznanych
Aby przekuć te spostrzeżenia w praktyczne przewidywania, zespół wytrenował nadzorowane modele uczenia maszynowego — w tym regresję logistyczną, lasy losowe oraz klasyfikator probabilistyczny — na fizykochemicznych odciskach palca kinaz o znanych etykietach. Po starannym dostrojeniu i walidacji krzyżowej modele te potrafiły precyzyjnie przypisać kinazy do głównych klas używając zaledwie kilku składowych głównych. Zastosowane do 66 kinaz pozostawionych w zbiorze „OTHER”, modele konsekwentnie przeklasyfikowały kilka z nich, takich jak CDC7 i członkowie rodziny ULK, do konkretnych klas kinaz. Kontrole strukturalne tych przeklasyfikowań, zwłaszcza wokół wcześniej zidentyfikowanych kluczowych pozycji, potwierdziły przewidywania maszyn i pokazały, jak metoda może kierować ponowną klasyfikacją oraz dalszymi badaniami eksperymentalnymi.
Ogólny przepis na mapowanie rodzin białkowych
Mówiąc prościej, praca ta pokazuje, że białka można sortować nie tylko przez spisywanie ich sekwencji, lecz także przez destylowanie tego, jak ich rdzeniowe części zachowują się fizycznie i chemicznie. Dla kinaz widok skoncentrowany na domenie i oparty na właściwościach odtwarza znane rodziny, pomaga przelabelować odstające przypadki i uwypukla strukturalne „gorące miejsca” ważne dla aktywności i regulacji. Ponieważ przepis opiera się jedynie na wspólnym wyrównaniu domeny i ogólnych deskryptorach aminokwasów, autorzy pokazują również, że można go zastosować do innych grup białek, takich jak małe GTPazy, i potencjalnie rozszerzyć na immunoglobuliny, receptory sprzężone z białkiem G i inne. W miarę jak takie mapy stają się coraz bardziej dopracowane, mogą kierować projektowaniem bardziej selektywnych leków, pomagać w interpretacji mutacji powodujących choroby i oferować wyraźniejszą, funkcjonalnie zorientowaną mapę wszechświata białek.
Cytowanie: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Słowa kluczowe: kinazy białkowe, klasyfikacja białek, uczenie maszynowe, domeny białkowe, biologia oparta na strukturze