Clear Sky Science · es
Novedoso método universal centrado en dominios para la clasificación de proteínas
Por qué ordenar las proteínas importa para la salud
Dentro de cada célula, miles de diminutas máquinas proteicas mantienen la vida en funcionamiento. Entre las más importantes están las cinasas de proteínas, enzimas que activan o desactivan a otras proteínas y que son objetivos principales de muchos fármacos modernos, en particular las terapias contra el cáncer. Pero los científicos todavía tienen dificultades para organizar de forma limpia todas las cinasas en familias que reflejen su modo de acción. Este artículo presenta una nueva forma de clasificar cinasas—y, en principio, muchas otras proteínas—centrándose en la región central compartida que realiza la función, y en las características físicas y químicas básicas de sus bloques constructores. Esto promete mapas más nítidos de las familias de proteínas y, en última instancia, mejores pistas para diseñar medicamentos.
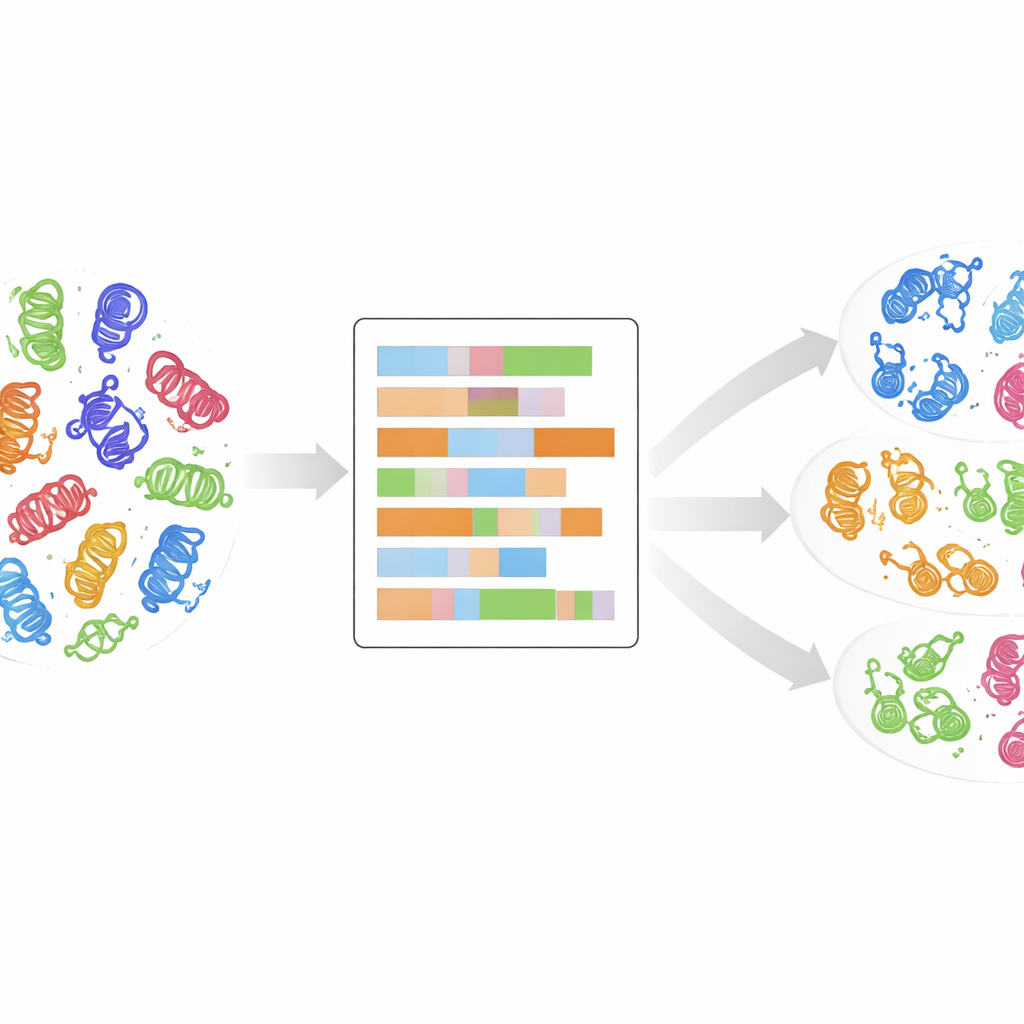
De los árboles genealógicos a una huella digital más detallada
Tradicionalmente, las cinasas se han agrupado en clases comparando sus secuencias de genes o proteínas y construyendo “árboles familiares” evolutivos. Este enfoque ha sido muy exitoso, revelando alrededor de 500 cinasas humanas que se agrupan en varios grupos principales como AGC, CAMK, CMGC, STE, TK, TKL y otros. Sin embargo, muchas cinasas recién descubiertas no encajan limpiamente en estos grupos: sus secuencias parecen distintas, aunque pueden comportarse de forma similar en la célula. Las comparaciones estándar también prestan poca atención a las propiedades subyacentes de cada aminoácido—el tamaño, la carga o la afinidad por el agua que determinan cómo se pliega y funciona una proteína. Los autores sostienen que para comprender realmente las familias de cinasas necesitamos mirar más allá del emparejamiento alfabético de secuencias y examinar estas huellas físico-químicas.
Acercándose al núcleo activo de las cinasas
Para ello, los investigadores utilizaron un alineamiento de alta calidad de 497 dominios de cinasas humanas, los núcleos compactos que realizan la reacción química de añadir grupos fosfato. Para cada posición en este mapa de dominio compartido, reemplazaron la letra del aminoácido por hasta 30 descriptores numéricos que capturan propiedades como carga, hidrofobicidad, polaridad y tamaño, además de un marcador extra para huecos. El resultado es un retrato numérico detallado de cada dominio de cinasa, donde un comportamiento similar en el espacio tridimensional debería traducirse en patrones similares en estos números. Luego redujeron la complejidad de estos retratos usando análisis de componentes principales, una técnica estándar que condensa muchas mediciones en unas pocas direcciones principales que capturan las mayores diferencias.
Dejando que los datos se agrupen por sí mismos
Sin decirle al ordenador a qué clase conocida pertenecía cada cinasa, el equipo aplicó un método de agrupamiento no supervisado llamado k-means a los datos numéricos reducidos. Exploraron muchos posibles números de clústeres y usaron puntuaciones estadísticas para identificar las agrupaciones más significativas, luego combinaron varias de estas soluciones en un conjunto final de 24 clústeres, cada uno con una puntuación de confianza que refleja su estabilidad entre ejecuciones. Sorprendentemente, alrededor del 90% de las cinasas terminó en clústeres que concordaban con sus etiquetas de clase originales, lo que sugiere que el retrato de dominio físico-químico recupera de forma natural—y a veces afina—las clasificaciones existentes. Algunos clústeres contenían una mezcla de una clase mayoritaria y cinasas previamente etiquetadas como “OTRAS”, lo que sugiere que esos casos atípicos podrían, de hecho, pertenecer a una familia establecida.
Descubriendo puntos calientes estructurales clave
Más allá del agrupamiento, el método revela qué partes del dominio de la cinasa impulsan realmente estas diferencias. Al combinar los componentes principales con las propiedades de los residuos y luego mezclar los datos en pruebas de aleatorización, los autores identificaron posiciones específicas cuyas pautas de propiedades distinguen con fuerza a una clase de las demás. Un ejemplo destacado es un sitio en la región del bucle de activación de las cinasas CMGC que casi siempre tiene un residuo cargado positivamente, a diferencia de la mayoría de las otras clases. Los modelos estructurales muestran que, en una cinasa representativa de CMGC, ese residuo ayuda a estabilizar sitios fosforilados cercanos que son cruciales para activar la enzima. Curiosamente, una cinasa “no clasificada” llamada CDC7 comparte un entorno similar en este sitio, lo que respalda la predicción de que se comporta como una cinasa CMGC aunque su historia evolutiva sea diferente.
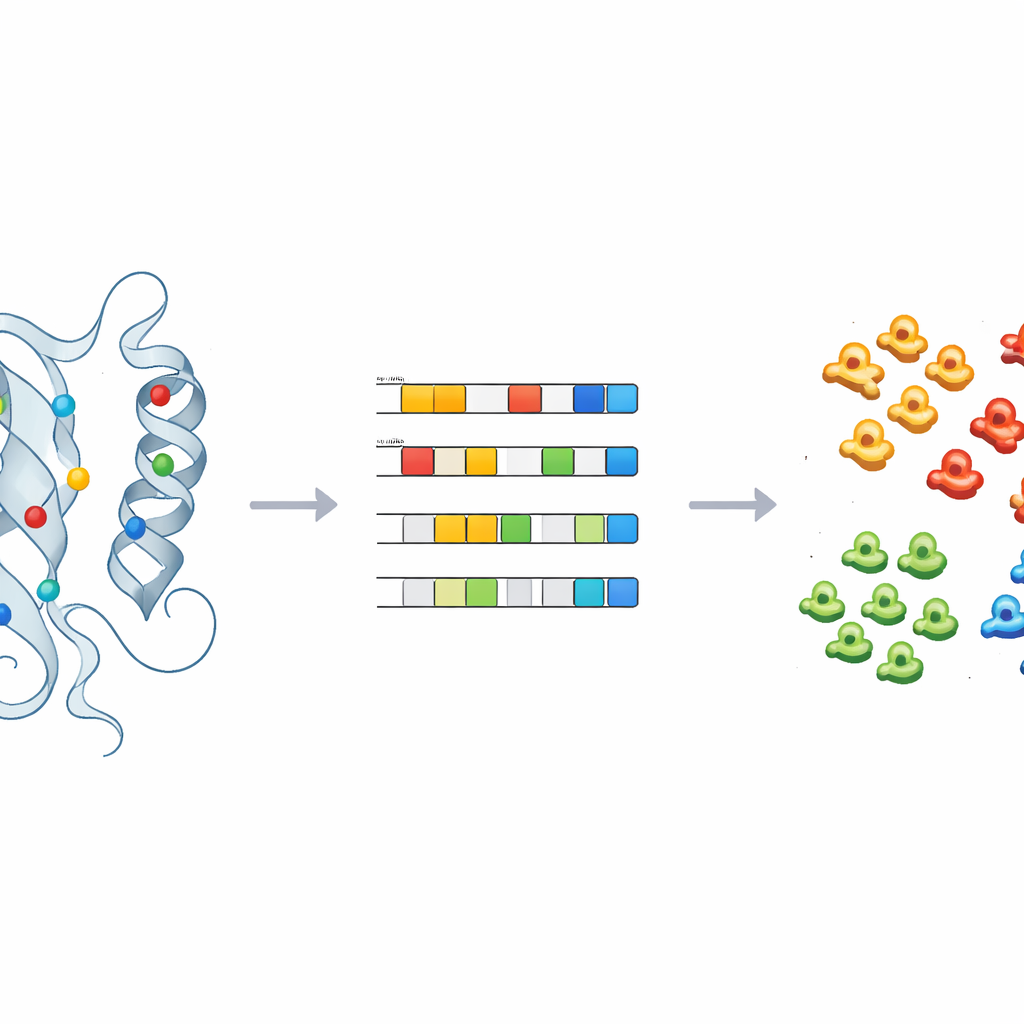
Enseñar a las máquinas a etiquetar lo desconocido
Para convertir estos conocimientos en predicciones prácticas, el equipo entrenó modelos supervisados de aprendizaje automático—incluyendo regresión logística, bosques aleatorios y un clasificador probabilístico—sobre las huellas físico-químicas de las cinasas con etiquetas conocidas. Tras un ajuste cuidadoso y validación cruzada, estos modelos pudieron asignar con precisión las cinasas a las clases principales usando solo un puñado de componentes principales. Aplicados a 66 cinasas que habían quedado en el grupo genérico “OTRAS”, los modelos reasignaron de forma consistente a varias, como CDC7 y miembros de la familia ULK, a clases de cinasas específicas. Verificaciones estructurales de estas reasignaciones, especialmente alrededor de las posiciones clave identificadas anteriormente, respaldaron las predicciones de las máquinas e ilustraron cómo el método puede guiar la reclasificación y el seguimiento experimental.
Una receta general para cartografiar familias de proteínas
En términos sencillos, este trabajo muestra que las proteínas pueden ordenarse no solo escribiendo sus secuencias, sino destilando cómo se comportan físicamente y químicamente sus partes centrales. Para las cinasas, esta perspectiva centrada en dominios y basada en propiedades recupera familias conocidas, ayuda a reetiquetar desviaciones y destaca “puntos calientes” estructurales que importan para la actividad y la regulación. Dado que la receta se basa únicamente en un alineamiento de dominio compartido y descriptores generales de aminoácidos, los autores también demuestran que puede aplicarse a otros grupos proteicos, como las pequeñas GTPasas, y podría extenderse a inmunoglobulinas, receptores acoplados a proteínas G y más. A medida que estos mapas se refinen, podrían orientar el diseño de fármacos más selectivos, ayudar a interpretar mutaciones que causan enfermedades y ofrecer un atlas más claro y centrado en la función del universo proteico.
Cita: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Palabras clave: cinasas de proteínas, clasificación de proteínas, aprendizaje automático, dominios proteicos, biología basada en estructura