Clear Sky Science · de
Neue universelle domänenzentrierte Methode zur Protein-Klassifikation
Warum die Einordnung von Proteinen für die Gesundheit wichtig ist
In jeder Zelle sorgen tausende winziger Proteinmaschinen dafür, dass das Leben reibungslos abläuft. Zu den wichtigsten gehören Proteinkinasen, Enzyme, die andere Proteine ein- oder ausschalten und daher Hauptziele vieler moderner Medikamente sind, insbesondere in der Krebstherapie. Trotzdem tun sich Wissenschaftler noch schwer, alle Kinasen sauber in Familien einzuordnen, die ihr Wirkungsprinzip widerspiegeln. Dieser Artikel stellt eine neue Methode vor, Kinasen — und prinzipiell viele andere Proteine — zu klassifizieren, indem er sich auf die gemeinsame Kernregion konzentriert, die die Funktion erfüllt, sowie auf die grundlegenden physikalisch-chemischen Eigenschaften ihrer Bausteine. Das verspricht schärfere Karten von Proteinfamilien und letztlich bessere Hinweise für die Wirkstoffentwicklung.
Von Stammbaumdarstellungen zu einem detaillierten Fingerabdruck
Traditionell wurden Kinasen durch den Vergleich ihrer Gen- oder Proteinsequenzen und den Aufbau evolutionärer „Stammbäume“ in Klassen gruppiert. Dieser Ansatz war sehr erfolgreich und identifizierte etwa 500 menschliche Kinasen, die in mehrere Hauptgruppen fallen, wie AGC, CAMK, CMGC, STE, TK, TKL und andere. Dennoch passen viele neu entdeckte Kinasen nicht sauber in diese Gruppen: Ihre Sequenzen sehen unterschiedlich aus, obwohl sie im Zellkontext ähnlich funktionieren können. Standardvergleiche berücksichtigen außerdem kaum die zugrunde liegenden Eigenschaften jeder Aminosäure — Größe, Ladung oder wasserliebender Charakter — die bestimmen, wie ein Protein faltet und arbeitet. Die Autoren argumentieren, dass wir, um Kinasefamilien wirklich zu verstehen, über alphabetähnliche Sequenzvergleiche hinausblicken und diese physiko-chemischen Fingerabdrücke untersuchen müssen.
Hineinzoomen auf den aktiven Kern der Kinasen
Um dies zu erreichen, nutzten die Forschenden eine hochwertige Ausrichtung von 497 menschlichen Kinase-Domänen, den kompakten Kernen, die tatsächlich die chemische Reaktion des Phosphatübertrags ausführen. Für jede Position in dieser gemeinsamen Domänenkarte ersetzten sie den Aminosäurebuchstaben durch bis zu 30 numerische Deskriptoren, die Eigenschaften wie Ladung, Hydrophobizität, Polarität und Größe abbilden, plus einen zusätzlichen Marker für Lücken. Das Ergebnis ist ein detailliertes numerisches Porträt jeder Kinase-Domäne, bei dem ähnliches dreidimensionales Verhalten sich in ähnlichen Mustern dieser Zahlen niederschlagen sollte. Anschließend reduzierten sie die Komplexität dieser Porträts mittels Hauptkomponentenanalyse, einer Standardtechnik, die viele Messgrößen auf wenige Hauptachsen kondensiert, welche die größten Unterschiede erfassen.
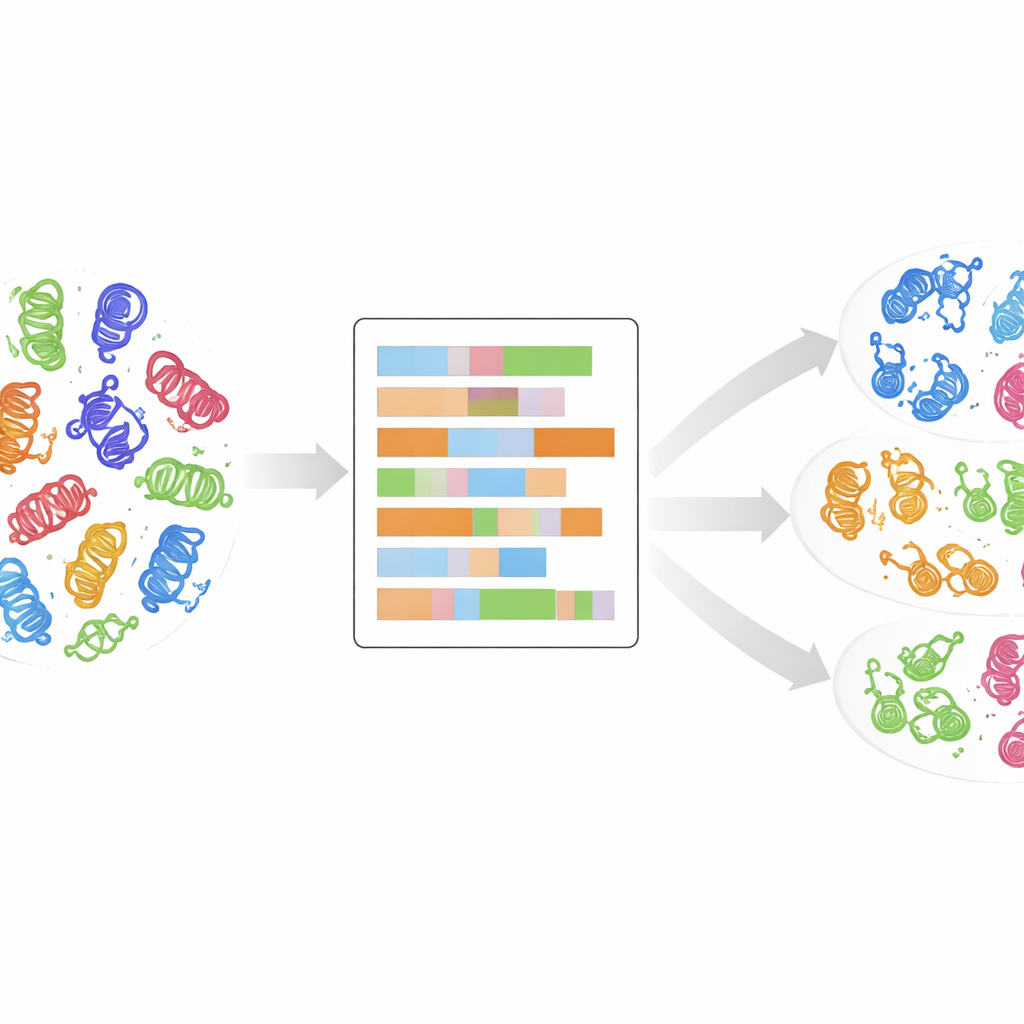
Die Daten sich selbst clustern lassen
Ohne dem Computer vorzugeben, welche Kinase zu welcher bekannten Klasse gehört, wandte das Team ein unüberwachtes Clustering-Verfahren namens k-means auf die reduzierten numerischen Daten an. Sie untersuchten viele mögliche Clusterzahlen und nutzten statistische Kennziffern, um die aussagekräftigsten Gruppierungen zu identifizieren, und kombinierten mehrere solcher Lösungen zu einer endgültigen Menge von 24 Clustern, jedes mit einem Konfidenzwert, der angibt, wie stabil es über verschiedene Läufe war. Bemerkenswerterweise landeten rund 90 % der Kinasen in Clustern, die mit ihren ursprünglichen Klassenlabels übereinstimmten, was darauf hindeutet, dass das physiko-chemische Domänenporträt bestehende Klassifikationen natürlicherweise wiederfindet — und manchmal sogar schärft. Einige Cluster enthielten eine Mischung aus einer Hauptklasse und zuvor als „OTHER“ eingestuften Kinasen, was darauf hinweist, dass diese Ausreißer tatsächlich zu einer etablierten Familie gehören könnten.
Wesentliche strukturelle Hotspots entdecken
Über die Gruppierung hinaus zeigt die Methode, welche Teile der Kinase-Domäne diese Unterschiede tatsächlich antreiben. Durch die Kombination der Hauptkomponenten mit den Resteigenschaften und anschließende Randomisierungs-Tests identifizierten die Autoren spezifische Positionen, deren Eigenschaftsmuster eine Klasse deutlich von den anderen unterscheiden. Ein herausragendes Beispiel ist eine Stelle in der Aktivierungsschleife der CMGC-Kinasen, die fast immer eine positiv geladene Aminosäure trägt, anders als die meisten anderen Klassen. Strukturmodelle zeigen, dass diese Aminosäure in einem repräsentativen CMGC-Kinaseenzym dazu beiträgt, benachbarte phosphorylierte Stellen zu stabilisieren, die für das Einschalten des Enzyms entscheidend sind. Interessanterweise teilt eine „unklassifizierte“ Kinase namens CDC7 eine ähnliche Umgebung an dieser Stelle, was die Vorhersage stützt, dass sie sich wie eine CMGC-Kinase verhält, auch wenn ihre evolutionäre Geschichte anders sein mag.
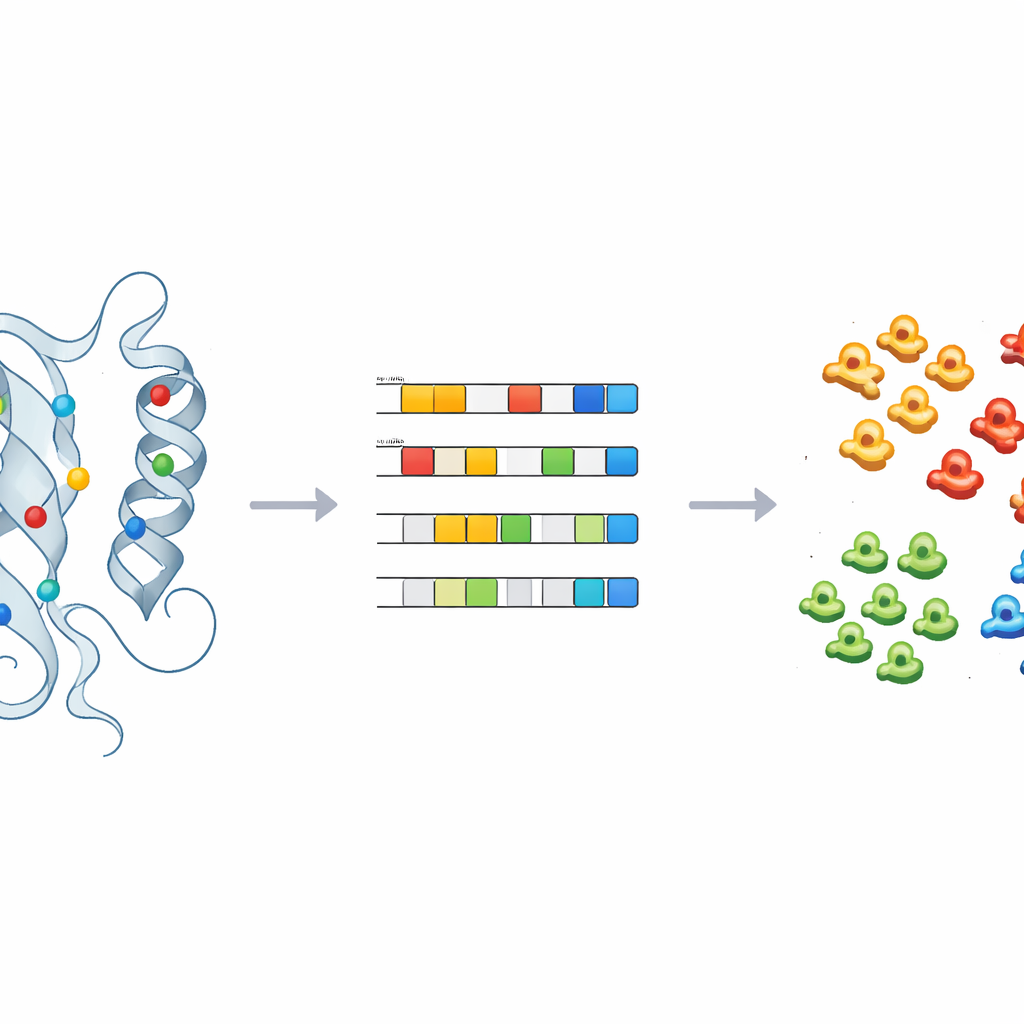
Maschinen beibringen, Unbekannte zu benennen
Um diese Einsichten in praktische Vorhersagen zu verwandeln, trainierte das Team überwachtes maschinelles Lernen — darunter logistische Regression, Random Forests und einen probabilistischen Klassifikator — auf den physiko-chemischen Fingerabdrücken von Kinasen mit bekannten Labels. Nach sorgfältiger Feinabstimmung und Kreuzvalidierung konnten diese Modelle Kinasen mit hoher Genauigkeit den Hauptklassen zuordnen, wobei nur wenige Hauptkomponenten verwendet wurden. Auf 66 Kinasen angewandt, die zuvor in der Sammelkategorie „OTHER“ verblieben waren, ordneten die Modelle mehrere konsequent neu zu, etwa CDC7 und Mitglieder der ULK-Familie, und wiesen sie spezifischen Kinaseklassen zu. Strukturchecks dieser Neuzuordnungen, insbesondere rund um die zuvor identifizierten Schlüsselpositionen, unterstützten die Maschinenvorhersagen und zeigten, wie die Methode Reklassifikation und experimentelle Nachuntersuchungen leiten kann.
Ein allgemeines Rezept zur Kartierung von Proteinfamilien
Alltäglich gesprochen zeigt diese Arbeit, dass Proteine nicht nur nach ihrer Sequenz „buchstabiert“ werden können, sondern dass man daraus destillieren kann, wie ihre Kernabschnitte sich physikalisch und chemisch verhalten. Für Kinasen stellt diese domänenzentrierte, eigenschaftsbasierte Sichtweise bekannte Familien wieder her, hilft Fehlzuordnungen zu korrigieren und hebt strukturelle „Hotspots“ hervor, die für Aktivität und Regulation wichtig sind. Da das Verfahren nur auf einer gemeinsamen Domänenausrichtung und allgemeinen Aminosäurebeschreibungen beruht, demonstrieren die Autoren außerdem, dass es auf andere Proteingruppen anwendbar ist, etwa kleine GTPasen, und potenziell auf Immunglobuline, G-Protein-gekoppelte Rezeptoren und darüber hinaus erweitert werden kann. Wenn solche Karten verfeinert werden, könnten sie die Entwicklung selektiverer Wirkstoffe lenken, bei der Interpretation krankheitsverursachender Mutationen helfen und einen klareren, funktionsorientierten Atlas des Proteinuniversums bieten.
Zitation: Fadaei, S., Krebs, F.S. & Zoete, V. Novel universal domain-centric method for protein classification. Sci Rep 16, 11850 (2026). https://doi.org/10.1038/s41598-026-41142-w
Schlüsselwörter: Proteinkinasen, Proteinklassifikation, maschinelles Lernen, Proteindomänen, strukturbasierte Biologie